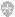单基因病的遗传方式
2011-11-17 18:20:24 来源: 作者: 评论:0 点击:
单基因遗传病简称单基因病(monogenic disease;single gene disorder)是指单一基因突变引起的疾病,符合孟德尔遗传方式,所以称为孟德尔式遗传病。
由于人类病症和性状不能如动物或植物那样通过杂交试验研究其遗传规律,因而必须采取适合于人类特点的研究方法。家系调查和系谱分析是判断某种遗传病遗传方式最常用的方法。系谱分析(pedigree analysis)是指将调查某患者家族成员所得到的该病或性状发生情况的资料,按一定格式绘制成图解(系谱)。对某病或性状遗传方式的判断必须进行多个系谱综合分析后方能作出准确结论。
绘制系谱图时采用统一的符号以表示家系中各个成员情况和相互之间的关系(图4-1)。
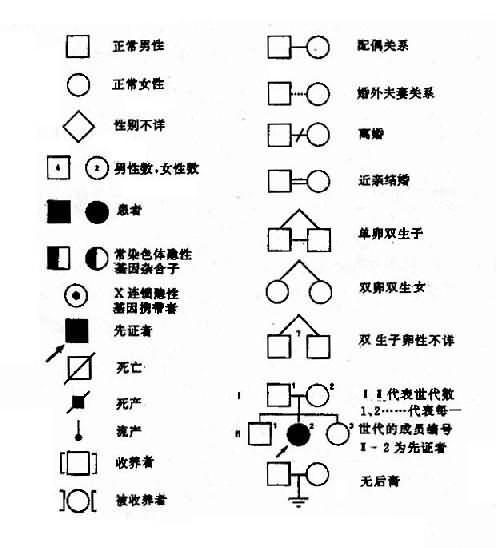
图4-1系谱符号
单基因病遗传方式的基本类型分述如下。
一、常染色体遗传
单基因涉及单个即一对等位基因发生突变所致的疾病,可按遗传方式分为下列4种主要类型。
1.常染色体显性遗传 一种遗传性状或遗传病有关的基因位于常染色体上,其性质是显性的,这种遗传方式称为常染色体显性遗传(autosomal dominant inheritance,AD)。由这种致病基因导致的疾病称为染色体显性遗传病。据统计,此类遗传病或异常性状已达3711种(1992年)。
致病基因有显性和隐性之分,其区别在于杂合状态(Aa)时,是否表现出相应的性或遗传病。若杂合子(Aa)能表现出与显性基因A有关的性状或遗传病时,其遗传方式称为显性遗传。
(1)完全显性遗传:凡是致病基因杂合状态(Aa)时,表现出像纯合子一样的显性性为状或遗传病者,称为完全显性(complete dominance)。短指症(brachydactyly)可作为完全显性遗传的实例。本症为较常见的手(足)部畸畸形。由于指骨或掌骨变短,或指骨缺如,致使手指(趾)变短(图4-2)。
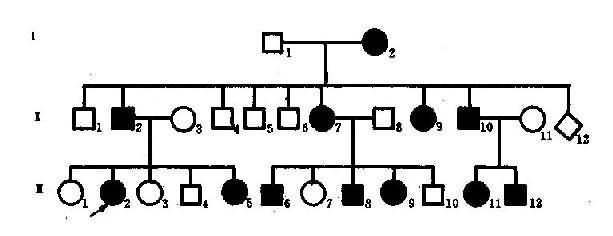
图4-2 一例短指(趾)症系谱
从系谱分析(图4-2),男女都可发病,与性别无关,所以本病是由某对常染色体上的基因决定的。假定A为显性基因(dominant gene),杂合状态时(Aa),只有基因A控制的性状显示出来,呈现出某种临床症状,而基因a的作用没有表达出来,称为隐性基因(recssive gene)。临床症状是表现出来的性状,称为表现型或表型(phenotype)。例如患者有短指,正常人没有短指,这是不同的表现型。控制各种表现型的遗传组成称为基因型或遗传型(genotype)。设短指症病人的基因型是Aa,正常人的基因型是aa.。等位基因Aa的人,两个基因不同,称为杂合子(heterozygote)。而一对基因相同(aa或AA)者,称为纯合子(homozygote)。因为A对a是显性的,基因A的作用在杂合子时表现出来,所以短指症的遗传方式是常染色体显性遗传。短指症的基因型有两种,纯合子(AA)和杂合子(Aa),它们在临床表现上无区别,故为完全显性。但临床上常见的情况都是杂合子患者(Aa)和正常人(aa)之间的婚配,后代中短指症患者与正常人的比例应为1:1,也就是说,后代将有约1/2子女发病,当两个短指症杂合子患者婚配时,其后代约3/4的子女将发病,只有约1/4子女正常。
图4-2的每个患者基因型都是杂合子(Aa),他(她)们的致病基因A一定来自双亲中的一方,所以双亲中的一方也是Aa,当然也是患者,这样就出现三代连续传递的现象。正常人的基因型都是aa,因此,患者的正常亲属也应都是aa,其子女都可能完全正常。该家系共21人,短指症患者11人(男5女6),发病比例接近1/2。应该指出,这种比例是在大样本的观察中方能反映出来,在子女数较少的小家庭往往不能反映出这种特点而出现较大的偏差。上述系谱基本反映了完全显性遗传特点,表现在:①连续四代发病;②患者子女中约1/2发病;③男女发病机会大致均等。
(2)不完全显性:有时杂合子(Aa)的表现型较纯合子轻,这种遗传方式称为不完全显性(incomplete dominance)或半显性(semi-dominance),也称中间型遗传(intermedeate inheritance)。这里,杂合子(Aa)中的显性基因A和隐性基因a的作用都得到-定程度的表达。β地中海贫血可作不完全显性遗传实例,致病基因βO纯合子,基因型为βOβO者病情严重,杂合子基因型为βOβA者病情较轻,而正常基因βA纯合子基因型为βAβA者无症状。从临床症状轻重来看,杂合子βOβA病情是界于βOβO与βAβA之间(详见本章第三节)。
(3)共显性:一对常染色体上的等位基因,彼此间没有显性和隐性的区别,在杂合状态时,两种基因都能表达,分别独立地产生基因产物,这种遗传方式称为共显性遗传(co-dominace)。ABO血型的遗传可作为显性遗传的实例。ABO血型决定于一组复等位基因(multiple alleles)。复等位基因是指在一个群体中,一对特定的基因座位上的基因不是两种(如A和a),而是三种或三种以上,有时可达数十种。但是,对每一个人来说只能具有其中的任何两个等位基因。复等位基因是由于一个基因发生多种突变,从而产生多种基因型的结果。ABO血型的基因已定位于9q34,在这一座位上,由IA、IB、和i三种基因组成复等位基因。基因IA对基因i为显性,基因IB对基因i也是显性。基因型IAIA和IAi都决定红细胞膜上抗原A的产生,这种个体为A型血;基因型IBIB和IBi都决定红细胞膜上抗原B的产生,这种个体为B型血;基因型ii测定H物质的产生而不产生抗原A和抗原B。就IA、IB、i这一组复等位来说,复等位基因的数目是3个,所以共有:n(n+1)/2=3(3+1)/2=6
种基因型。在共显性时,有4种表现型。如果纯合子(IAIA)A型血的人与纯合子(IBIB)B型血的人结婚只能出生杂合子(IAIB)AB型血的子女;如果两个杂合子(IAIB)AB型血的人结婚则会导致1(IAIA):2(IAIB):1(IBIB)的比率,这样,3:1的比值就被1:2:1的比值所代替,这是两个等位基因共显性的结果。
根据孟德尔分离律的原理,已知双亲血型,就可以估计出子女中可能出现的血型和不可能出现的血型(表4-1),这在法医学的亲子鉴定中有一定意义。
表4-1双亲和子女之间血型遗传的关系
双亲的血型子女中可能出现的血型子女中不可能出现的血型
A×AA,OB,AB
A×0A,OB,AB
A×BA,B,AB,O-
A×ABA,B,ABO
B×BB,OA,AB
B×OB,OA,AB
B×ABA,A,ABO
AB×OA,BAB,O
AB×ABA,B,ABO
O×OOA,B,AB
此外,人类MN血型、人类组织相容性抗原(human lecocyte antigen,HLA)系统等都是共显性例子。
(4)不规则显性:带有显性基因的个体理应发病,但事实上并非完全如此,有些杂合子(Aa)并不发病,这可能是因受修饰基因等因素的影响而不表现出临床症状,失去显性特点而不外显,有时表现程度有差异,称为不规则显性(irregular dominance)。修饰基因(modifier gene)是指本身没有表型效应,可是能对主基因发生影响,使主基因的表型成完全或能削弱主基因的作用,从而出现各种表现度和不完全的外显率。
图4-3 一例成骨不全病例系谱
图4-3是一个成骨不全症的系谱,该家族中的患者有一共同的致病基因(A),均同I1传递而来,然而他们的临床表现却有很大的差别。先证者Ⅲ8有蓝色巩膜和多次骨折,其母亲Ⅱ5只有一次骨折史,其姨母Ⅱ3和外祖母Ⅰ1都只有蓝色巩膜,其姨表兄Ⅲ5则具多次骨折和耳聋两种症状,可见这些患者存在着明显的表现程度不一致。杂合子(Aa)在不同遗传背景和环境因素影响下,性状表现程度的差异称为表现度(expressivity)。上述种患者症状表现速度的区别可以解释为:Ⅰ1.Ⅱ3由于遗传背景中可能存在着减弱基因(reducer gene)作用,所以表现度轻:Ⅲ4 、Ⅲ5和Ⅲ8的遗传背景中可能存在增强基因(enhancer gene),所以表现度重。
(5)延迟显性:有些显性遗传病并非出生后即表现出来,而是到较晚期才出现症状,这种情况称为延迟显性(delayed dominance)。慢性进行性舞蹈病(Huntington’s chorea)可作为实例。此病为染色体显性遗传病,致病基因位于4p16。杂合子(Aa)在20岁时只有1%发病,40岁有38%发病,60岁有94%发病。这里,年龄对发病是一个重要的修饰因素。可见本病杂合子要个体发育早期,致病基因并不表达,但到一定年龄后,致病基因的作用方表达出来,主称为延迟显性。
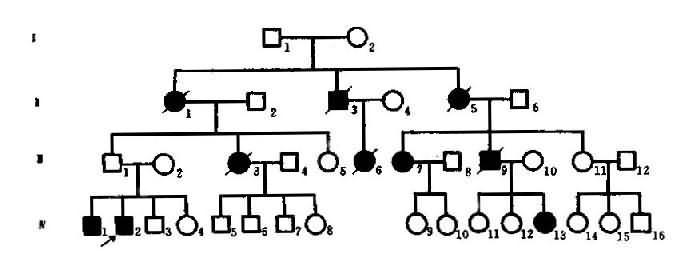
图4-4 例慢性进行舞蹈病系谱
图4-4是一例慢性进行性舞蹈病系谱。Ⅲ1未见发病,但他的母亲(Ⅱ1)和2个儿子(Ⅲ1,2)均已患病,因此可以认为Ⅲ1携带有致病基因,由于某种原因未能表现症状,因而出现了隔代传递现象。显性基因完全不能表达的个体称顿挫型(form fruste)。Ⅲ1是顿挫型,虽未发病的,便仍将致病基因传给后代。因此,本例是不规则显性。顿挫型的存在形成致病基因(A)不完全外显,这样显性基因在杂全状态时是否得到表现,可用外显就绪来衡量。外显率(penetrance)是指显性基因能形成相应表现型的比例,一般用百分率(%)来表示。显性基因能100%表现出相应性状称为完全外显,外显率低于100%时为不完全外显或外显不全。一般外显率高者可达70%-80%,低者只有20%-30%。当计算外显率时应搜集较多的家系汇总分析方能符合实际情况。
延迟显性的一特点是,最年轻一代的患者比例常不足1/2。图4-4中的第Ⅳ代患者仅3/13即为佐证。
2.常染色体隐性遗传控制遗传性状或遗传病的基因位于常染色体上,其性质是隐性的,在杂合状态时不表现相应性状,只有当隐性基因纯合子(aa)方得以表现,称为常为常染色体隐性遗传病(autosomal recessive inheritance,AR)。这种致病基因所引起的疾病称为常染色体隐性遗传病。目前已知的常染色体隐性遗传病或异常性状达1631种(1992年)。白化病(albinism)可作为常染色体隐性遗传病的实例。白化病是由于全身黑色素细胞均缺乏黑色素,所以皮肤毛发呈白色。本病患者只有当一对等位基因是隐性致病基因纯合子(aa)时才发病,所以患者的基因型都是纯合子(aa)。当一个个体为杂合状态(Aa)时,虽然本人不发病,但为致病的基因携带者,他(她)能将致病基因a传给后代,因此患者父母双方都应是致病基因(Aa)的肯定携带者(obligatory carrier)。如果两个杂合子(Aa)婚配,后代子女患者(aa)占1/4,表型正常者占3/4。表型正常的人中1/3基因型为纯合子(AA),2/3为杂合子(Aa),是致病基因的可能携带者(probable carrier)。
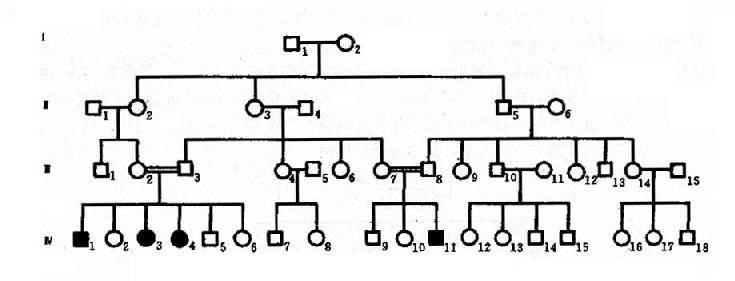
图4-5 一例白化病系谱
图4-5是白化病的一个家系,这个系谱基本反映了常染色体隐性遗传的特点。表现在:①患者(Ⅳ1,3,4,11)的双亲(Ⅲ2,3和Ⅲ7,8)表现型正常,但均为致病基因的肯定携带者;②系谱中看不到连续遗传现象,常为散发,有的系谱中只见先证者;③同胞中约1/4个体发病,男女性发病机会均等,患者大部分出现在同胞之间,子女往往正常;④近亲婚配的后代发病概率显著较高,系谱中的Ⅲ2和Ⅲ3.Ⅲ7和Ⅲ8都是近亲婚配。
二、性染色体遗传
性染色体上的基因所控制的遗传性状或遗传病,在遗传上总是和性别相关的。目前已知的性连锁遗传的致病基因大都在X染色体上,与性别相关联的遗传方式称为性连锁遗传(sexlinked inheritance)。目前已知的X连锁隐性遗传病或异常性状有360种(1992年)。
1.X连锁隐性遗传 一种性状或遗传病有关的基因位于X染色体上,这些基因的性质是隐性的,并随着X染色体的行为而传递,其遗传方式称为X连锁隐性遗传(X-linked recessive inheritance,XR).
以隐性方式遗传时,由于女性有两条X染色体,当隐性致病基因在杂合状态(XAXa)时,隐性基因控制的性状或遗传病不显示出来,这样的女性表型正常的致病基因携带者。只有当两条X染色体上等位基因都是隐性致病基因纯合子(XaXa)时才表现出来。在男性细胞中,只有一条X染色体,Y染色体上缺少同源节段,所以只要X染色体上有一个隐性致病基因(XaY)就发病。这样,男性的细胞中只有成对的等位基因中的一个基因,故称为半合子(hemizygote)。
红绿色盲可作X连锁隐性遗传病实例。色盲有全色盲(achromatopsis)和红色绿色盲(dyschromatopsia of the protan and deutan)之分。前者不能辨别任何颜色,一般认为是常染色体隐性遗传;后者最为常见,表现为对红绿色的辨别力降低。呈X连锁隐性遗传,致病基因定位于Xq28。据报道,男性发生率7.0%,女性为0.5%.一个红绿色盲男患者(XbY)和正常辩色能力女性(XBXB)结婚,他们的女儿都应从父亲那里接受一个X染色体,从母亲那里得到一条正常的X染色体而成为致病基因携带者杂合了(XBXb),他们的儿子必定由母亲那里接受一条XB,故辩色能力全部正常(XBY)。凡携带致病基因的女性(XBXb)与正常辩色男人结婚,下一代中,儿子有一半是正常(XBY)的,一半是绿色盲(XbY),女儿中一半是致病基因携带者(XBXb),一半则完全正常(XBXB)。因此,男性患者的父亲一定是患者,其母亲是致病基因携带者。这里可见“父传女,母传子”的交叉遗传(criss-cross inheritance)现象,如果女性携带者(XBXb)与男性患者(XbY)s结婚,后代中,女儿1/2可能发病,1/2可能为携带乾,独生子中发病者和正常者各占1/2。
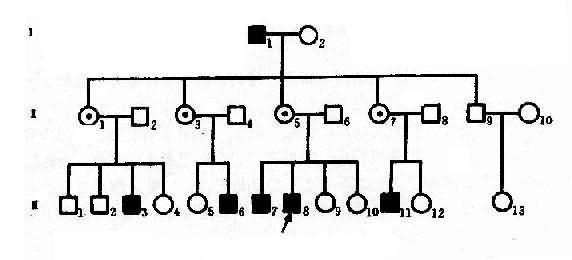
图4-6 一例红绿色盲系谱
从图4-6红绿色盲系谱分析,男性红绿色盲患者I1(XbY)和正常辩色能力女性I2结婚,他们的女儿全部是杂合子(Ⅱ1,3,5,7),因为儿了(Ⅱ9)只能从母亲处得到XB,故表型正常,在下一代中,携带致病基因的女性(XBXb)与正常男性(XBY)结婚时,他们的儿子有1/2可能是正常的(XBY),1/2可能是红绿色盲(XbY)(Ⅲ3,6,7,8,11),女儿中1/2可能是携带者(XBXb),1/2可能完全正常(XBXB),这样出现代与代间明显的间隔遗传现象。该系谱先证者Ⅲ8的妹妹Ⅲ9,10姨表姐妹Ⅲ4,5,12,-14虽表型正常,但有1/2可能是携带者,她们结婚后也有可能把致病基因传给儿子。
从红绿色盲系谱中,可反映出X连锁隐性遗传系谱和特点,表现在:①男性患者过远多于女性患者,系谱中的病人几乎都是男性;②男性患者的双亲都无病,其致病基因来自携带者母亲;③由于交叉遗传,男患者的同胞、舅父、姨表兄弟、外甥中常见到患者,偶见外祖父发病,在此情况下,男患者的舅父一般正常;④由于男患者的子女都是正常的,所以代与代间可见明显的不连续(隔代遗传)。
2.X连锁显性遗传 一些性状或遗传病的基因位于X染色体上,其性质是显性的,这种遗传方式称为X连锁显性遗传(X-linked dominant inheritance),这种疾病称为X连锁显性遗传病。目前所知X连锁显性遗传病不足20种。
由于致病基因是显性的,并位于X染色体上,因此,不论男性(XAY)和女性(XAXa)只要有一个这种致病基因XA就会发病。与常染色体显性遗传不同之处是,女性患者既可将致病基因传给生子,又可以传给女儿,且机会均等;而男性患者只能将致病基因传给女儿,不传给儿子。由此可见,女性患者多于男性,大约为男性的1倍。另外,从临床上看,女性患者大多数是杂合子,病情一般较男性轻,而男患者病情较重。
抗维生素D佝偻病(vitamin D resistant rickets, VDRR)可以作为X连锁显性遗传病的实例。VDRR是一种以低磷酸血症导致骨发育障碍为特征的遗传性骨病。患者主要是肾远曲小管对磷的转运机制有某种障碍,困而尿排磷酸盐增多,血磷酸盐降低而影响骨质钙化。患者身体矮小,有时伴有佝偻病等各种表现。患者用常规剂量的维生素D治疗不能奏效,故有抗维生素D佝偻病之称。从临床观察,女性患者的病情较男性患者轻,多数只有低血磷,佝偻症状不太明显,表现为不完全显性,这可能是女性患者多为杂合子,其中正常X染色体的基因还发挥一定的作用。
男性患者(XHY)与正常女性(XhXh)结婚,所生子女中,儿子全部正常,女儿全部发病;女性患者(XHXh)与正常男性(XhX)结婚,子女中正常与患者各占1/2。
图4-7是抗维生素D佝偻病系谱,女性患者I1(XHXh)产生两种配子,她与正常男性结婚,理论上子女正常与患者各占1/2,故Ⅱ1、Ⅱ2、Ⅱ3都可能发病,Ⅱ3的子女Ⅲ12,13,14也可能发病;但男性患者与正常女性结婚,由于男性患者把致病基因只传给他的女儿,不传给儿子,所以Ⅲ12和Ⅲ14的女儿(Ⅳ18,Ⅳ21)都发病,儿子(Ⅳ19,Ⅳ20)正常。同时可见到上代传给下代的连续性。女性(Ⅲ3、4、6)患者只有低血磷,没有佝偻病。本系谱可以反映出X连锁显性遗传特点,表现在:①女性患者多于男性,女性患者(Ⅲ3、4、6)病情较轻,只有低血磷;②患者双亲之一必定是患者,女患者都是杂合子,她们的致病基因可传给儿子和女儿,但男患者的致病基因只传给女儿,因此系谱中男患者的女儿全部发病;③可看到连续两代以上都有患者。
3.Y连锁遗传 如果致病基因位于Y染色体上,并随着Y染色体而传递,故只有男性才出现症状。这类致病基因只由父亲传给儿子,再由儿子传给孙子,女性是不会出现相应的遗传性状或遗传病,这种遗传方式称为Y连锁遗传(Y-linked inheritance)。由于这些基因控制的性状,只能在雄性个体中表现,这种现象又称为限雄遗传(holandric inheritance)。
图4-7 一例抗维生素D佝偻病系谱
迄今报道Y连锁遗传病及异常性状仅10余种。我国发现一个视网膜色素变性的家系,4代共26人中,8例患者均为男性,女性正常且后代亦无患者,很可能属Y连锁遗传,有待进一步证实。另外耳毛性状呈Y连锁遗传较多见。
除上述几种基本遗传方式外,尚有2种特殊情况:
(1)从性遗传:从性遗传睡性连锁遗传的表现都与性别有密切关系,但它们是两种截然不同的遗传方式。性连锁遗传的基因位于性染色体上,而从性遗传的基因位于常染色体上,致病基因性质有显性和隐性之别。这种常染色体上的基因所控制的性状,在表现型上受性别影响而男女性分布比例或表现程度上的差别,这种遗传方式称为从性遗传(sex-influrenced inheritance)。
原发性血色病(primary hematochromatosis)可作从性遗传方式的实例。本病为一种遗传性铁代谢障碍,其特征为含铁血黄素在组织中大量沉积,造成多种器官损害,典型症状是皮肤色素沉着、肝硬化、糖尿病三联综合征,症状发生较迟,由于铁质蓄积达到15-30g方产生症状,所以80%病例在40岁以后发病。本病致病基因在常染色体上,但男性多于女性10-20倍,而且女性发病较迟,这是因为女性通过月经、妊娠和哺乳,一生顺可丧失铁10-35g,故难以表现铁质沉着症状。
遗传性早秃(hereditary alopecia)为常染色体显性遗传病,男性显著多于女性,女性仅表现为头发稀疏,极少全秃,杂合子(Bb)男性会出现早秃;相反,女性杂合子(Bb)不出现早秃,只有纯合子(BB)才出现早秃,这也是从性遗传的一例。
(2)限性遗传;一种遗传性状或遗传病的致病基因位于常染色体或性染色体上,其性质可以是显性或隐性,但由于性别限制,只在一种性别得以表现,而在另一性别完全不能表现,但这些基因都可以向后代传递,这种遗传方式称为限性遗传(sex-limited inheritance)。例如,子宫阴道积水(hydrometrocolpos)由常染色体隐性基因决定,因此,女性只有在纯合子才表现相应症状,男性虽有这种基因但不能表现该性状,然而这些基因都向后代传递。
上述从性遗传和限性遗传特点可见,并非所有表现出性别差异的遗传性状或遗传病都是性连锁遗传,在常染色体遗传病中有时也可见到性别差异,应注意加以区别。
三、两种单基因病或性状的遗传规律
1.两种单基因病的致病基因分别位于不同对染色体上,在临床上,一个家系中如果出现两种单基因病患者,在大多数情况下,其遗传方式受孟德尔自由组合律制约。假设一个并指症(并指完全,伴有掌、跖骨融合)女性与一个甲型血友男患者结婚,生育了一个男孩,其并指症状与母亲类同,他们要求再生一孩子,试问后代子女发病风险如何?
并指症是常染色体性遗传病,因此母亲的基因型为XXBb,甲型血友病是X连锁隐性遗传病,男性是半合子发病,所以父亲的基因型是XhYbb。从图4-8可以看出,他们后代的女孩中,有50%可能患并指症伴甲型血友病携带者,50%可能是甲型血友病携带者;男孩中,50%可能是并指症,但有50%可能正常。
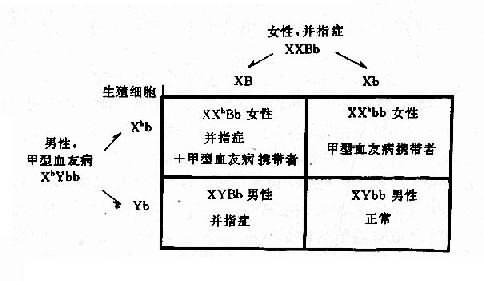
图4-8 两种致病基因在不同染色体上的自由组合
B:并指症基因;h:甲型血友病基因
2.两种单基因病基因位于同一染色体上 有时患者有两种单基因遗传病,如果这两种致病基因位于同一染色体上,它们将表现为连锁遗传,其遗传方式受连锁与交换律制约。例如,红绿色盲与甲型血友病的基因都是在X染色体上,所以彼此连锁。假定两者间交换率是10%。如果父亲是红绿色盲,母亲外表正常,已生出一个女儿是红绿色盲,一个儿子是甲型血友病,试问他们以后所生的孩子中,这两种遗传病的发病风险如何?能生出正常的后代吗?
从一个女儿是红绿色盲来看,母亲必然是红绿色盲基因(b)的携带者,再从一个儿子是甲型血友病来看,母亲也必然是甲型血友病基因(h)的携带者。但是,这两种致病基因分别位于两条染色上。父亲是红绿色盲患者,所以有红绿色盲基因(b)。从图4-9可以看出,他们后代的女孩中,50%可能是正常的,50%可能患红绿色盲;男孩中,45%可能患红绿色盲,45%可能患甲型血友病,5%可能同时患这两种病,只有5%可能是正常的。
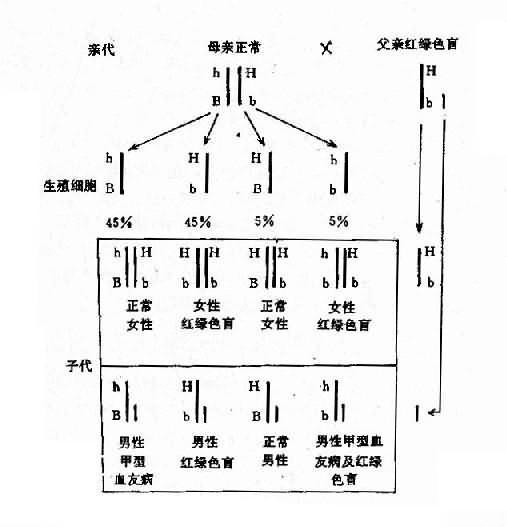
图4-9 两种X连锁隐性遗传病的连锁和交换
h:甲型血友病基因:b:红绿色盲基因
综上所述,研究两种基因病伴随遗传规律,在遗传咨询时估测遗传病患者后代发病风险是重要的。
3.两对基因的相互作用 先天性聋哑是一个较常见的两对基因相互作用的例子。假定在不同座位上的两套以上的隐性基因中,只要任何一个座位是隐性纯合子,就出现聋哑。
(1)聋哑双亲生育的子女可以是全部聋哑或全部正常
DDee×DDee→DDee
ddEE×ddEE→ddEE
DDee×ddEE→DdEe 全部正常
(2)不同家系的聋哑尊重亲生育正常听觉子代相互结婚,有较高机会出现聋哑儿女
即正常听觉占9/16,聋哑占7/16
四、单基因病的遗传异质性与遗传方式
遗传异质性(heterogeneity)是指表现型一致的个体或同种疾病临床表现相同,但可能具不同的基因型,称为遗传异质性。由于遗传基础不同,它们的遗传方式、发病年龄、病程进展、病情严重程度、预后以及复发风险等都可能不同。研究表明,遗传病病种增多的原因不仅是由于发现了新的疾病,而是从已知的综合征中分出了亚型,即遗传异质性的存在。遗传异质性几乎成为遗传的普通现象。例如视网膜色素变性(retinitis pigmentosa,RP)是最常见的致盲的单基因遗传眼病之一,主要表现为视网膜萎缩、夜盲和视野缩小,多为双眼发病,致中年或老年进完全失明。
RP的遗传方式具有遗传异质性,即可以有AD、AR、XR连锁遗传,可能还有Y连锁遗传。遗传方式不同的RP,一般其遗传基础也不同,因而伴随的综合征的以及始发年龄、主要病情变化特征(XR常伴高度近视,AR和AD多为低度近视)、病程进展(AD快,AR慢)、预后情况(AD较轻,AR致盲)也有差异,甚至还可区分为其他不同亚型。
现知,XL的RP2基因定位于Xp11.4-11.23,XL的PR3基因定位于Xp21.1,AD的RP基因定位于8p11-q21。因此,普遍认为RP是多个基因座位上RP基因所引起的一组具有临床亚型的视网膜退行性病变的遗传性疾病。
五、不同于孟德尔遗传规律的遗传现象
1.母系遗传 母系遗传(maternal inheritance)是指核外染色体所控制的遗传现象。例如Leber遗传性视神经病(Leber’s heredi tary optic neuropathy,LHON),也称Leber病。其主要病变为视神经退行性变,发病较早,表现为急性亚急性视力减退,中心视野丧失最明显。此病发病机制一般认为是由于mtDNA点突变导致其第11778位精氨酸→组氨酸(多见)及细胞色素b第15257位天冬氨酸→天冬酰胺。前者使编码呼吸链NADH脱氢酶mtDNA第340位精氨酸被组氨酸取代,改变了mtDNA阀间构型,导致NADH脱氢酶活性降低,线粒体产能下降,因而对需能量多的视神经组织损害最大,久之导致视神经细胞退行性变,直至萎缩。
由于mtDNA为母系遗传,因此由mtDNA基因突变所致的Leber病也遵循母系遗传的传递规律,即患者都与母亲有关。男性患者的后代中尚未见有直接传代者。但并非女性患者的后代全部发病,而且发病年龄也不一致;甚至一些女性患者本身表型正常,但可将本病传给下一代。母系遗传的特点:①母亲将她的mtDNA传递给儿子和女儿,但只有女儿能将其mtDNA传递给下一代;②人的细胞里通常有上千个mtDNA拷贝,在突变体和正常mtDNA共存的细胞中,mtDNA在细胞的复制和分离过程中发生遗传漂变,可导致子细胞出现三种基因型:纯合的突变体mtDNA、纯合的正常mtDNA、突变体和正常的mtDNA的杂合,这是由于mtDNA的遗传不遵循孟德尔定律,被随机分配到子细胞中所致;线粒体病发病有一阈值,只有当异常的mtDNA超过阈值时才发病。女性携带者的细胞内突变的mtDNA未达到阈值或在某种程度上受核影响而未发病,但仍可以通过mtDNA突变体向下代传递。女性患者细胞里mtDNA同样可能存在杂合性,子女中得到较多突变mtDNA的个体发病,得到较少的病情较轻或不发病。
2.遗传印记根据孟德尔的遗传定律,当一个性状从亲本传给子代,无论携带这个性状的基因或染色体来自父方或母方,所产生的表型效应是相同的。但是目前发现同一种染色体(或基因)的改变由于不同性别的亲本传给子女时可以引起不同的疾病。例如,Prader-Willi综合征(PWS)和Angelman综合征(AS)是两种不同的遗传病,但都有共同的15q11-13缺失。父源染色体缺失时临床上为PWS,而母源染色体缺失时表现为AS。这提示来源不同的等位基因有不同的表达。某些常染色体显性遗传病的发病年龄和病情轻重似乎与传递基因亲本有关。慢性进行性舞蹈病患者发病年龄一般在30-50岁,但有5%-10%患者在20岁以前发病,且病情严重,这些患者致病基因均由父亲遗传。母亲遗传者,子女发病年龄多在40-50岁。囊性纤维化(cystic fibrosis,CF)是一种常染色体隐性遗传病,已发现某些CF患者的二条7号染色体均来自母亲,即单亲二体性(uniparental disomy,UPD)。人类的胚胎发育也有类似现象,拥有父源两套染色体的受精卵发育成葡萄胎,而拥有母源两套染色体的发育成卵巢畸胎瘤。此外,无论是双雄三倍体还是双雌三倍体都发育成畸胎儿。因此,正常的胚胎发育必须拥有亲代双方染色体或基因组。一些胚胎性肿瘤中也存在亲源性非随机的染色体或基因丢失现象,而且主要是母源染色体的丢失。如散发的肾母细胞瘤(Wilms trmor)有11p13-15的基因丢失,且皆来自母方,而遗传型基因丢失多来自父方;遗传型视网膜母细胞瘤(Rb)中有13q14杂合性丢失(LOH),且丢失的多为母系源Rb基因(详第九章)。
目前已知,至少有数十种遗传病存在着遗传印记现象,这种现象很难用经典的孟德尔定律来解释,也不能用性连锁遗传、线粒体遗传及多基因遗传来回答。近年来,揭示了一种新的遗传现象,即基因组印记(genomic imprinting),亦称遗传印记(genetic imprinting),是指来自双亲的基因或染色体存在着功能上的差异,因而子女来自父方与来自母方的基因表达可以不同。这是由于基因在生殖细胞分化过程中受到不同修饰的结果。换言之,遗传印记是一种依赖于配子起源的某些等位基因的修饰现象.一些基因在精子生成过程中被印记,另一些基因在卵子生成过程中被印记,被印记了的基因,它们的表达受到抑制。
遗传印记现象已在哺乳动物和人类中确认,但对印记现象的机理仍了解很少。据推测DNA的甲基化可能遗传印记的分子机理之一。在精子和卵子中一些基因甲基化程度不同,高度甲基化(被印记)的基因不表达或表达程度降低,当胚胎发育过程中发生去甲基化时,这些基因即开始表达。总之,基因的印记影响到性状或许多遗传病和肿瘤的发生,影响发病年龄、外显率、表现度,甚至遗传方式。在对某些不能用经典孟德尔定律解释的遗传现象时,用遗传印记可以得到合理解释。
由于人类病症和性状不能如动物或植物那样通过杂交试验研究其遗传规律,因而必须采取适合于人类特点的研究方法。家系调查和系谱分析是判断某种遗传病遗传方式最常用的方法。系谱分析(pedigree analysis)是指将调查某患者家族成员所得到的该病或性状发生情况的资料,按一定格式绘制成图解(系谱)。对某病或性状遗传方式的判断必须进行多个系谱综合分析后方能作出准确结论。
绘制系谱图时采用统一的符号以表示家系中各个成员情况和相互之间的关系(图4-1)。
图4-1系谱符号
单基因病遗传方式的基本类型分述如下。
一、常染色体遗传
单基因涉及单个即一对等位基因发生突变所致的疾病,可按遗传方式分为下列4种主要类型。
1.常染色体显性遗传 一种遗传性状或遗传病有关的基因位于常染色体上,其性质是显性的,这种遗传方式称为常染色体显性遗传(autosomal dominant inheritance,AD)。由这种致病基因导致的疾病称为染色体显性遗传病。据统计,此类遗传病或异常性状已达3711种(1992年)。
致病基因有显性和隐性之分,其区别在于杂合状态(Aa)时,是否表现出相应的性或遗传病。若杂合子(Aa)能表现出与显性基因A有关的性状或遗传病时,其遗传方式称为显性遗传。
(1)完全显性遗传:凡是致病基因杂合状态(Aa)时,表现出像纯合子一样的显性性为状或遗传病者,称为完全显性(complete dominance)。短指症(brachydactyly)可作为完全显性遗传的实例。本症为较常见的手(足)部畸畸形。由于指骨或掌骨变短,或指骨缺如,致使手指(趾)变短(图4-2)。
图4-2 一例短指(趾)症系谱
从系谱分析(图4-2),男女都可发病,与性别无关,所以本病是由某对常染色体上的基因决定的。假定A为显性基因(dominant gene),杂合状态时(Aa),只有基因A控制的性状显示出来,呈现出某种临床症状,而基因a的作用没有表达出来,称为隐性基因(recssive gene)。临床症状是表现出来的性状,称为表现型或表型(phenotype)。例如患者有短指,正常人没有短指,这是不同的表现型。控制各种表现型的遗传组成称为基因型或遗传型(genotype)。设短指症病人的基因型是Aa,正常人的基因型是aa.。等位基因Aa的人,两个基因不同,称为杂合子(heterozygote)。而一对基因相同(aa或AA)者,称为纯合子(homozygote)。因为A对a是显性的,基因A的作用在杂合子时表现出来,所以短指症的遗传方式是常染色体显性遗传。短指症的基因型有两种,纯合子(AA)和杂合子(Aa),它们在临床表现上无区别,故为完全显性。但临床上常见的情况都是杂合子患者(Aa)和正常人(aa)之间的婚配,后代中短指症患者与正常人的比例应为1:1,也就是说,后代将有约1/2子女发病,当两个短指症杂合子患者婚配时,其后代约3/4的子女将发病,只有约1/4子女正常。
图4-2的每个患者基因型都是杂合子(Aa),他(她)们的致病基因A一定来自双亲中的一方,所以双亲中的一方也是Aa,当然也是患者,这样就出现三代连续传递的现象。正常人的基因型都是aa,因此,患者的正常亲属也应都是aa,其子女都可能完全正常。该家系共21人,短指症患者11人(男5女6),发病比例接近1/2。应该指出,这种比例是在大样本的观察中方能反映出来,在子女数较少的小家庭往往不能反映出这种特点而出现较大的偏差。上述系谱基本反映了完全显性遗传特点,表现在:①连续四代发病;②患者子女中约1/2发病;③男女发病机会大致均等。
(2)不完全显性:有时杂合子(Aa)的表现型较纯合子轻,这种遗传方式称为不完全显性(incomplete dominance)或半显性(semi-dominance),也称中间型遗传(intermedeate inheritance)。这里,杂合子(Aa)中的显性基因A和隐性基因a的作用都得到-定程度的表达。β地中海贫血可作不完全显性遗传实例,致病基因βO纯合子,基因型为βOβO者病情严重,杂合子基因型为βOβA者病情较轻,而正常基因βA纯合子基因型为βAβA者无症状。从临床症状轻重来看,杂合子βOβA病情是界于βOβO与βAβA之间(详见本章第三节)。
(3)共显性:一对常染色体上的等位基因,彼此间没有显性和隐性的区别,在杂合状态时,两种基因都能表达,分别独立地产生基因产物,这种遗传方式称为共显性遗传(co-dominace)。ABO血型的遗传可作为显性遗传的实例。ABO血型决定于一组复等位基因(multiple alleles)。复等位基因是指在一个群体中,一对特定的基因座位上的基因不是两种(如A和a),而是三种或三种以上,有时可达数十种。但是,对每一个人来说只能具有其中的任何两个等位基因。复等位基因是由于一个基因发生多种突变,从而产生多种基因型的结果。ABO血型的基因已定位于9q34,在这一座位上,由IA、IB、和i三种基因组成复等位基因。基因IA对基因i为显性,基因IB对基因i也是显性。基因型IAIA和IAi都决定红细胞膜上抗原A的产生,这种个体为A型血;基因型IBIB和IBi都决定红细胞膜上抗原B的产生,这种个体为B型血;基因型ii测定H物质的产生而不产生抗原A和抗原B。就IA、IB、i这一组复等位来说,复等位基因的数目是3个,所以共有:n(n+1)/2=3(3+1)/2=6
种基因型。在共显性时,有4种表现型。如果纯合子(IAIA)A型血的人与纯合子(IBIB)B型血的人结婚只能出生杂合子(IAIB)AB型血的子女;如果两个杂合子(IAIB)AB型血的人结婚则会导致1(IAIA):2(IAIB):1(IBIB)的比率,这样,3:1的比值就被1:2:1的比值所代替,这是两个等位基因共显性的结果。
根据孟德尔分离律的原理,已知双亲血型,就可以估计出子女中可能出现的血型和不可能出现的血型(表4-1),这在法医学的亲子鉴定中有一定意义。
表4-1双亲和子女之间血型遗传的关系
双亲的血型子女中可能出现的血型子女中不可能出现的血型
A×AA,OB,AB
A×0A,OB,AB
A×BA,B,AB,O-
A×ABA,B,ABO
B×BB,OA,AB
B×OB,OA,AB
B×ABA,A,ABO
AB×OA,BAB,O
AB×ABA,B,ABO
O×OOA,B,AB
此外,人类MN血型、人类组织相容性抗原(human lecocyte antigen,HLA)系统等都是共显性例子。
(4)不规则显性:带有显性基因的个体理应发病,但事实上并非完全如此,有些杂合子(Aa)并不发病,这可能是因受修饰基因等因素的影响而不表现出临床症状,失去显性特点而不外显,有时表现程度有差异,称为不规则显性(irregular dominance)。修饰基因(modifier gene)是指本身没有表型效应,可是能对主基因发生影响,使主基因的表型成完全或能削弱主基因的作用,从而出现各种表现度和不完全的外显率。
图4-3 一例成骨不全病例系谱
图4-3是一个成骨不全症的系谱,该家族中的患者有一共同的致病基因(A),均同I1传递而来,然而他们的临床表现却有很大的差别。先证者Ⅲ8有蓝色巩膜和多次骨折,其母亲Ⅱ5只有一次骨折史,其姨母Ⅱ3和外祖母Ⅰ1都只有蓝色巩膜,其姨表兄Ⅲ5则具多次骨折和耳聋两种症状,可见这些患者存在着明显的表现程度不一致。杂合子(Aa)在不同遗传背景和环境因素影响下,性状表现程度的差异称为表现度(expressivity)。上述种患者症状表现速度的区别可以解释为:Ⅰ1.Ⅱ3由于遗传背景中可能存在着减弱基因(reducer gene)作用,所以表现度轻:Ⅲ4 、Ⅲ5和Ⅲ8的遗传背景中可能存在增强基因(enhancer gene),所以表现度重。
(5)延迟显性:有些显性遗传病并非出生后即表现出来,而是到较晚期才出现症状,这种情况称为延迟显性(delayed dominance)。慢性进行性舞蹈病(Huntington’s chorea)可作为实例。此病为染色体显性遗传病,致病基因位于4p16。杂合子(Aa)在20岁时只有1%发病,40岁有38%发病,60岁有94%发病。这里,年龄对发病是一个重要的修饰因素。可见本病杂合子要个体发育早期,致病基因并不表达,但到一定年龄后,致病基因的作用方表达出来,主称为延迟显性。
图4-4 例慢性进行舞蹈病系谱
图4-4是一例慢性进行性舞蹈病系谱。Ⅲ1未见发病,但他的母亲(Ⅱ1)和2个儿子(Ⅲ1,2)均已患病,因此可以认为Ⅲ1携带有致病基因,由于某种原因未能表现症状,因而出现了隔代传递现象。显性基因完全不能表达的个体称顿挫型(form fruste)。Ⅲ1是顿挫型,虽未发病的,便仍将致病基因传给后代。因此,本例是不规则显性。顿挫型的存在形成致病基因(A)不完全外显,这样显性基因在杂全状态时是否得到表现,可用外显就绪来衡量。外显率(penetrance)是指显性基因能形成相应表现型的比例,一般用百分率(%)来表示。显性基因能100%表现出相应性状称为完全外显,外显率低于100%时为不完全外显或外显不全。一般外显率高者可达70%-80%,低者只有20%-30%。当计算外显率时应搜集较多的家系汇总分析方能符合实际情况。
延迟显性的一特点是,最年轻一代的患者比例常不足1/2。图4-4中的第Ⅳ代患者仅3/13即为佐证。
2.常染色体隐性遗传控制遗传性状或遗传病的基因位于常染色体上,其性质是隐性的,在杂合状态时不表现相应性状,只有当隐性基因纯合子(aa)方得以表现,称为常为常染色体隐性遗传病(autosomal recessive inheritance,AR)。这种致病基因所引起的疾病称为常染色体隐性遗传病。目前已知的常染色体隐性遗传病或异常性状达1631种(1992年)。白化病(albinism)可作为常染色体隐性遗传病的实例。白化病是由于全身黑色素细胞均缺乏黑色素,所以皮肤毛发呈白色。本病患者只有当一对等位基因是隐性致病基因纯合子(aa)时才发病,所以患者的基因型都是纯合子(aa)。当一个个体为杂合状态(Aa)时,虽然本人不发病,但为致病的基因携带者,他(她)能将致病基因a传给后代,因此患者父母双方都应是致病基因(Aa)的肯定携带者(obligatory carrier)。如果两个杂合子(Aa)婚配,后代子女患者(aa)占1/4,表型正常者占3/4。表型正常的人中1/3基因型为纯合子(AA),2/3为杂合子(Aa),是致病基因的可能携带者(probable carrier)。
图4-5 一例白化病系谱
图4-5是白化病的一个家系,这个系谱基本反映了常染色体隐性遗传的特点。表现在:①患者(Ⅳ1,3,4,11)的双亲(Ⅲ2,3和Ⅲ7,8)表现型正常,但均为致病基因的肯定携带者;②系谱中看不到连续遗传现象,常为散发,有的系谱中只见先证者;③同胞中约1/4个体发病,男女性发病机会均等,患者大部分出现在同胞之间,子女往往正常;④近亲婚配的后代发病概率显著较高,系谱中的Ⅲ2和Ⅲ3.Ⅲ7和Ⅲ8都是近亲婚配。
二、性染色体遗传
性染色体上的基因所控制的遗传性状或遗传病,在遗传上总是和性别相关的。目前已知的性连锁遗传的致病基因大都在X染色体上,与性别相关联的遗传方式称为性连锁遗传(sexlinked inheritance)。目前已知的X连锁隐性遗传病或异常性状有360种(1992年)。
1.X连锁隐性遗传 一种性状或遗传病有关的基因位于X染色体上,这些基因的性质是隐性的,并随着X染色体的行为而传递,其遗传方式称为X连锁隐性遗传(X-linked recessive inheritance,XR).
以隐性方式遗传时,由于女性有两条X染色体,当隐性致病基因在杂合状态(XAXa)时,隐性基因控制的性状或遗传病不显示出来,这样的女性表型正常的致病基因携带者。只有当两条X染色体上等位基因都是隐性致病基因纯合子(XaXa)时才表现出来。在男性细胞中,只有一条X染色体,Y染色体上缺少同源节段,所以只要X染色体上有一个隐性致病基因(XaY)就发病。这样,男性的细胞中只有成对的等位基因中的一个基因,故称为半合子(hemizygote)。
红绿色盲可作X连锁隐性遗传病实例。色盲有全色盲(achromatopsis)和红色绿色盲(dyschromatopsia of the protan and deutan)之分。前者不能辨别任何颜色,一般认为是常染色体隐性遗传;后者最为常见,表现为对红绿色的辨别力降低。呈X连锁隐性遗传,致病基因定位于Xq28。据报道,男性发生率7.0%,女性为0.5%.一个红绿色盲男患者(XbY)和正常辩色能力女性(XBXB)结婚,他们的女儿都应从父亲那里接受一个X染色体,从母亲那里得到一条正常的X染色体而成为致病基因携带者杂合了(XBXb),他们的儿子必定由母亲那里接受一条XB,故辩色能力全部正常(XBY)。凡携带致病基因的女性(XBXb)与正常辩色男人结婚,下一代中,儿子有一半是正常(XBY)的,一半是绿色盲(XbY),女儿中一半是致病基因携带者(XBXb),一半则完全正常(XBXB)。因此,男性患者的父亲一定是患者,其母亲是致病基因携带者。这里可见“父传女,母传子”的交叉遗传(criss-cross inheritance)现象,如果女性携带者(XBXb)与男性患者(XbY)s结婚,后代中,女儿1/2可能发病,1/2可能为携带乾,独生子中发病者和正常者各占1/2。
图4-6 一例红绿色盲系谱
从图4-6红绿色盲系谱分析,男性红绿色盲患者I1(XbY)和正常辩色能力女性I2结婚,他们的女儿全部是杂合子(Ⅱ1,3,5,7),因为儿了(Ⅱ9)只能从母亲处得到XB,故表型正常,在下一代中,携带致病基因的女性(XBXb)与正常男性(XBY)结婚时,他们的儿子有1/2可能是正常的(XBY),1/2可能是红绿色盲(XbY)(Ⅲ3,6,7,8,11),女儿中1/2可能是携带者(XBXb),1/2可能完全正常(XBXB),这样出现代与代间明显的间隔遗传现象。该系谱先证者Ⅲ8的妹妹Ⅲ9,10姨表姐妹Ⅲ4,5,12,-14虽表型正常,但有1/2可能是携带者,她们结婚后也有可能把致病基因传给儿子。
从红绿色盲系谱中,可反映出X连锁隐性遗传系谱和特点,表现在:①男性患者过远多于女性患者,系谱中的病人几乎都是男性;②男性患者的双亲都无病,其致病基因来自携带者母亲;③由于交叉遗传,男患者的同胞、舅父、姨表兄弟、外甥中常见到患者,偶见外祖父发病,在此情况下,男患者的舅父一般正常;④由于男患者的子女都是正常的,所以代与代间可见明显的不连续(隔代遗传)。
2.X连锁显性遗传 一些性状或遗传病的基因位于X染色体上,其性质是显性的,这种遗传方式称为X连锁显性遗传(X-linked dominant inheritance),这种疾病称为X连锁显性遗传病。目前所知X连锁显性遗传病不足20种。
由于致病基因是显性的,并位于X染色体上,因此,不论男性(XAY)和女性(XAXa)只要有一个这种致病基因XA就会发病。与常染色体显性遗传不同之处是,女性患者既可将致病基因传给生子,又可以传给女儿,且机会均等;而男性患者只能将致病基因传给女儿,不传给儿子。由此可见,女性患者多于男性,大约为男性的1倍。另外,从临床上看,女性患者大多数是杂合子,病情一般较男性轻,而男患者病情较重。
抗维生素D佝偻病(vitamin D resistant rickets, VDRR)可以作为X连锁显性遗传病的实例。VDRR是一种以低磷酸血症导致骨发育障碍为特征的遗传性骨病。患者主要是肾远曲小管对磷的转运机制有某种障碍,困而尿排磷酸盐增多,血磷酸盐降低而影响骨质钙化。患者身体矮小,有时伴有佝偻病等各种表现。患者用常规剂量的维生素D治疗不能奏效,故有抗维生素D佝偻病之称。从临床观察,女性患者的病情较男性患者轻,多数只有低血磷,佝偻症状不太明显,表现为不完全显性,这可能是女性患者多为杂合子,其中正常X染色体的基因还发挥一定的作用。
男性患者(XHY)与正常女性(XhXh)结婚,所生子女中,儿子全部正常,女儿全部发病;女性患者(XHXh)与正常男性(XhX)结婚,子女中正常与患者各占1/2。
图4-7是抗维生素D佝偻病系谱,女性患者I1(XHXh)产生两种配子,她与正常男性结婚,理论上子女正常与患者各占1/2,故Ⅱ1、Ⅱ2、Ⅱ3都可能发病,Ⅱ3的子女Ⅲ12,13,14也可能发病;但男性患者与正常女性结婚,由于男性患者把致病基因只传给他的女儿,不传给儿子,所以Ⅲ12和Ⅲ14的女儿(Ⅳ18,Ⅳ21)都发病,儿子(Ⅳ19,Ⅳ20)正常。同时可见到上代传给下代的连续性。女性(Ⅲ3、4、6)患者只有低血磷,没有佝偻病。本系谱可以反映出X连锁显性遗传特点,表现在:①女性患者多于男性,女性患者(Ⅲ3、4、6)病情较轻,只有低血磷;②患者双亲之一必定是患者,女患者都是杂合子,她们的致病基因可传给儿子和女儿,但男患者的致病基因只传给女儿,因此系谱中男患者的女儿全部发病;③可看到连续两代以上都有患者。
3.Y连锁遗传 如果致病基因位于Y染色体上,并随着Y染色体而传递,故只有男性才出现症状。这类致病基因只由父亲传给儿子,再由儿子传给孙子,女性是不会出现相应的遗传性状或遗传病,这种遗传方式称为Y连锁遗传(Y-linked inheritance)。由于这些基因控制的性状,只能在雄性个体中表现,这种现象又称为限雄遗传(holandric inheritance)。
图4-7 一例抗维生素D佝偻病系谱
迄今报道Y连锁遗传病及异常性状仅10余种。我国发现一个视网膜色素变性的家系,4代共26人中,8例患者均为男性,女性正常且后代亦无患者,很可能属Y连锁遗传,有待进一步证实。另外耳毛性状呈Y连锁遗传较多见。
除上述几种基本遗传方式外,尚有2种特殊情况:
(1)从性遗传:从性遗传睡性连锁遗传的表现都与性别有密切关系,但它们是两种截然不同的遗传方式。性连锁遗传的基因位于性染色体上,而从性遗传的基因位于常染色体上,致病基因性质有显性和隐性之别。这种常染色体上的基因所控制的性状,在表现型上受性别影响而男女性分布比例或表现程度上的差别,这种遗传方式称为从性遗传(sex-influrenced inheritance)。
原发性血色病(primary hematochromatosis)可作从性遗传方式的实例。本病为一种遗传性铁代谢障碍,其特征为含铁血黄素在组织中大量沉积,造成多种器官损害,典型症状是皮肤色素沉着、肝硬化、糖尿病三联综合征,症状发生较迟,由于铁质蓄积达到15-30g方产生症状,所以80%病例在40岁以后发病。本病致病基因在常染色体上,但男性多于女性10-20倍,而且女性发病较迟,这是因为女性通过月经、妊娠和哺乳,一生顺可丧失铁10-35g,故难以表现铁质沉着症状。
遗传性早秃(hereditary alopecia)为常染色体显性遗传病,男性显著多于女性,女性仅表现为头发稀疏,极少全秃,杂合子(Bb)男性会出现早秃;相反,女性杂合子(Bb)不出现早秃,只有纯合子(BB)才出现早秃,这也是从性遗传的一例。
(2)限性遗传;一种遗传性状或遗传病的致病基因位于常染色体或性染色体上,其性质可以是显性或隐性,但由于性别限制,只在一种性别得以表现,而在另一性别完全不能表现,但这些基因都可以向后代传递,这种遗传方式称为限性遗传(sex-limited inheritance)。例如,子宫阴道积水(hydrometrocolpos)由常染色体隐性基因决定,因此,女性只有在纯合子才表现相应症状,男性虽有这种基因但不能表现该性状,然而这些基因都向后代传递。
上述从性遗传和限性遗传特点可见,并非所有表现出性别差异的遗传性状或遗传病都是性连锁遗传,在常染色体遗传病中有时也可见到性别差异,应注意加以区别。
三、两种单基因病或性状的遗传规律
1.两种单基因病的致病基因分别位于不同对染色体上,在临床上,一个家系中如果出现两种单基因病患者,在大多数情况下,其遗传方式受孟德尔自由组合律制约。假设一个并指症(并指完全,伴有掌、跖骨融合)女性与一个甲型血友男患者结婚,生育了一个男孩,其并指症状与母亲类同,他们要求再生一孩子,试问后代子女发病风险如何?
并指症是常染色体性遗传病,因此母亲的基因型为XXBb,甲型血友病是X连锁隐性遗传病,男性是半合子发病,所以父亲的基因型是XhYbb。从图4-8可以看出,他们后代的女孩中,有50%可能患并指症伴甲型血友病携带者,50%可能是甲型血友病携带者;男孩中,50%可能是并指症,但有50%可能正常。
图4-8 两种致病基因在不同染色体上的自由组合
B:并指症基因;h:甲型血友病基因
2.两种单基因病基因位于同一染色体上 有时患者有两种单基因遗传病,如果这两种致病基因位于同一染色体上,它们将表现为连锁遗传,其遗传方式受连锁与交换律制约。例如,红绿色盲与甲型血友病的基因都是在X染色体上,所以彼此连锁。假定两者间交换率是10%。如果父亲是红绿色盲,母亲外表正常,已生出一个女儿是红绿色盲,一个儿子是甲型血友病,试问他们以后所生的孩子中,这两种遗传病的发病风险如何?能生出正常的后代吗?
从一个女儿是红绿色盲来看,母亲必然是红绿色盲基因(b)的携带者,再从一个儿子是甲型血友病来看,母亲也必然是甲型血友病基因(h)的携带者。但是,这两种致病基因分别位于两条染色上。父亲是红绿色盲患者,所以有红绿色盲基因(b)。从图4-9可以看出,他们后代的女孩中,50%可能是正常的,50%可能患红绿色盲;男孩中,45%可能患红绿色盲,45%可能患甲型血友病,5%可能同时患这两种病,只有5%可能是正常的。
图4-9 两种X连锁隐性遗传病的连锁和交换
h:甲型血友病基因:b:红绿色盲基因
综上所述,研究两种基因病伴随遗传规律,在遗传咨询时估测遗传病患者后代发病风险是重要的。
3.两对基因的相互作用 先天性聋哑是一个较常见的两对基因相互作用的例子。假定在不同座位上的两套以上的隐性基因中,只要任何一个座位是隐性纯合子,就出现聋哑。
(1)聋哑双亲生育的子女可以是全部聋哑或全部正常
DDee×DDee→DDee
ddEE×ddEE→ddEE
DDee×ddEE→DdEe 全部正常
(2)不同家系的聋哑尊重亲生育正常听觉子代相互结婚,有较高机会出现聋哑儿女
即正常听觉占9/16,聋哑占7/16
四、单基因病的遗传异质性与遗传方式
遗传异质性(heterogeneity)是指表现型一致的个体或同种疾病临床表现相同,但可能具不同的基因型,称为遗传异质性。由于遗传基础不同,它们的遗传方式、发病年龄、病程进展、病情严重程度、预后以及复发风险等都可能不同。研究表明,遗传病病种增多的原因不仅是由于发现了新的疾病,而是从已知的综合征中分出了亚型,即遗传异质性的存在。遗传异质性几乎成为遗传的普通现象。例如视网膜色素变性(retinitis pigmentosa,RP)是最常见的致盲的单基因遗传眼病之一,主要表现为视网膜萎缩、夜盲和视野缩小,多为双眼发病,致中年或老年进完全失明。
RP的遗传方式具有遗传异质性,即可以有AD、AR、XR连锁遗传,可能还有Y连锁遗传。遗传方式不同的RP,一般其遗传基础也不同,因而伴随的综合征的以及始发年龄、主要病情变化特征(XR常伴高度近视,AR和AD多为低度近视)、病程进展(AD快,AR慢)、预后情况(AD较轻,AR致盲)也有差异,甚至还可区分为其他不同亚型。
现知,XL的RP2基因定位于Xp11.4-11.23,XL的PR3基因定位于Xp21.1,AD的RP基因定位于8p11-q21。因此,普遍认为RP是多个基因座位上RP基因所引起的一组具有临床亚型的视网膜退行性病变的遗传性疾病。
五、不同于孟德尔遗传规律的遗传现象
1.母系遗传 母系遗传(maternal inheritance)是指核外染色体所控制的遗传现象。例如Leber遗传性视神经病(Leber’s heredi tary optic neuropathy,LHON),也称Leber病。其主要病变为视神经退行性变,发病较早,表现为急性亚急性视力减退,中心视野丧失最明显。此病发病机制一般认为是由于mtDNA点突变导致其第11778位精氨酸→组氨酸(多见)及细胞色素b第15257位天冬氨酸→天冬酰胺。前者使编码呼吸链NADH脱氢酶mtDNA第340位精氨酸被组氨酸取代,改变了mtDNA阀间构型,导致NADH脱氢酶活性降低,线粒体产能下降,因而对需能量多的视神经组织损害最大,久之导致视神经细胞退行性变,直至萎缩。
由于mtDNA为母系遗传,因此由mtDNA基因突变所致的Leber病也遵循母系遗传的传递规律,即患者都与母亲有关。男性患者的后代中尚未见有直接传代者。但并非女性患者的后代全部发病,而且发病年龄也不一致;甚至一些女性患者本身表型正常,但可将本病传给下一代。母系遗传的特点:①母亲将她的mtDNA传递给儿子和女儿,但只有女儿能将其mtDNA传递给下一代;②人的细胞里通常有上千个mtDNA拷贝,在突变体和正常mtDNA共存的细胞中,mtDNA在细胞的复制和分离过程中发生遗传漂变,可导致子细胞出现三种基因型:纯合的突变体mtDNA、纯合的正常mtDNA、突变体和正常的mtDNA的杂合,这是由于mtDNA的遗传不遵循孟德尔定律,被随机分配到子细胞中所致;线粒体病发病有一阈值,只有当异常的mtDNA超过阈值时才发病。女性携带者的细胞内突变的mtDNA未达到阈值或在某种程度上受核影响而未发病,但仍可以通过mtDNA突变体向下代传递。女性患者细胞里mtDNA同样可能存在杂合性,子女中得到较多突变mtDNA的个体发病,得到较少的病情较轻或不发病。
2.遗传印记根据孟德尔的遗传定律,当一个性状从亲本传给子代,无论携带这个性状的基因或染色体来自父方或母方,所产生的表型效应是相同的。但是目前发现同一种染色体(或基因)的改变由于不同性别的亲本传给子女时可以引起不同的疾病。例如,Prader-Willi综合征(PWS)和Angelman综合征(AS)是两种不同的遗传病,但都有共同的15q11-13缺失。父源染色体缺失时临床上为PWS,而母源染色体缺失时表现为AS。这提示来源不同的等位基因有不同的表达。某些常染色体显性遗传病的发病年龄和病情轻重似乎与传递基因亲本有关。慢性进行性舞蹈病患者发病年龄一般在30-50岁,但有5%-10%患者在20岁以前发病,且病情严重,这些患者致病基因均由父亲遗传。母亲遗传者,子女发病年龄多在40-50岁。囊性纤维化(cystic fibrosis,CF)是一种常染色体隐性遗传病,已发现某些CF患者的二条7号染色体均来自母亲,即单亲二体性(uniparental disomy,UPD)。人类的胚胎发育也有类似现象,拥有父源两套染色体的受精卵发育成葡萄胎,而拥有母源两套染色体的发育成卵巢畸胎瘤。此外,无论是双雄三倍体还是双雌三倍体都发育成畸胎儿。因此,正常的胚胎发育必须拥有亲代双方染色体或基因组。一些胚胎性肿瘤中也存在亲源性非随机的染色体或基因丢失现象,而且主要是母源染色体的丢失。如散发的肾母细胞瘤(Wilms trmor)有11p13-15的基因丢失,且皆来自母方,而遗传型基因丢失多来自父方;遗传型视网膜母细胞瘤(Rb)中有13q14杂合性丢失(LOH),且丢失的多为母系源Rb基因(详第九章)。
目前已知,至少有数十种遗传病存在着遗传印记现象,这种现象很难用经典的孟德尔定律来解释,也不能用性连锁遗传、线粒体遗传及多基因遗传来回答。近年来,揭示了一种新的遗传现象,即基因组印记(genomic imprinting),亦称遗传印记(genetic imprinting),是指来自双亲的基因或染色体存在着功能上的差异,因而子女来自父方与来自母方的基因表达可以不同。这是由于基因在生殖细胞分化过程中受到不同修饰的结果。换言之,遗传印记是一种依赖于配子起源的某些等位基因的修饰现象.一些基因在精子生成过程中被印记,另一些基因在卵子生成过程中被印记,被印记了的基因,它们的表达受到抑制。
遗传印记现象已在哺乳动物和人类中确认,但对印记现象的机理仍了解很少。据推测DNA的甲基化可能遗传印记的分子机理之一。在精子和卵子中一些基因甲基化程度不同,高度甲基化(被印记)的基因不表达或表达程度降低,当胚胎发育过程中发生去甲基化时,这些基因即开始表达。总之,基因的印记影响到性状或许多遗传病和肿瘤的发生,影响发病年龄、外显率、表现度,甚至遗传方式。在对某些不能用经典孟德尔定律解释的遗传现象时,用遗传印记可以得到合理解释。
相关热词搜索:
上一篇:基因突变致蛋白质合成异常
下一篇:基因突变










论坛新帖
医学推广
频道本月排行
热门购物
评论排行
- 2011年临床执业医师考试实践技能真...(13)
- 腋臭手术视频(11)
- 2008年考研英语真题及参考答案(5)
- 节食挑食最伤女人的免疫系统(5)
- 核辐射的定义和单位(5)
- CKD患者Tm与IMT相关(5)
- 齐鲁医院普外科开展“喉返神经监护...(5)
- windows7激活工具WIN7 Activation v1.7(5)
- 正常微循环(5)
- 美大学性教育课来真的 男女上阵亲...(4)