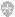遗传病
2011-10-21 17:25:32 来源: 作者: 评论:0 点击:
遣传性疾病通常具有垂直传递的特征,可由亲代传至后代。有时突变发生在配子形成(gametogenesis)时期,因此父母双方都没有这类缺陷,这类患者成为起始性突变者,可能成为后代子孙患病的祖先。有些基因突变或染色体畸变是致死性的或明显降低生殖力,以致这种缺陷不能传递。多基因型传递的遗传疾病,可能经长期一再地与正常个体婚配,而冲淡突变基因甚至消失。遗传性疾病常具有先天性、终生性和家族性的特点。但先天性疾病并不都是遗传病,在胎儿发育过程中,由于环境因素或母体条件的影响,可出现非遗传性先天性疾病。遗传性疾病也常表现为晚发,一些致病基因的作用仅在个体发育达到一定年龄后才表现出来。尤其是那些由遗传基因和环境条件两种因素支配着表现型的遗传性疾病。因此,晚发的疾病仍可以是遗传性疾病。遗传性疾病常可表现为家族性,这是因为同一家系中的成员可共同具有某一致病基因,但同一家系的成员也处于相似的生活条件和环境中,由相似环境条件所引起的非遗传性疾病,有时也具有家族性。一些遗传性疾病(隐性遗传病)仅在基因型为纯合子状态下才表现出来,因而发病的概率小,相似于散发性疾病。
作为遗传病的发病基础,近年来的认识越加深入。已往知之较多的是染色体畸变,畸变处出现染色体遗传物质的得失、断裂或位置改变而使机体出现病状。另一方面是基因突变,是编码基因的碱基序列发生变化,随之表达和表达产物异常。这种基因突变引起的疾病,随该基因所在染色体连锁、交换,按孟德尔的自由组合及分离律传递给下一代,称为孟德尔式遗传病。人类线粒体DNA是细胞核外DNA,它的DNA突变也可致线粒体功能异常出现疾病,总称线粒体遗传病。线粒体遗传病是由卵细胞胞浆内突变的线粒体DNA传递给下一代的。因此表现为母系遗传。家系中可见男女均可患病,但男患者后代再无患者。近年来人类基因研究发现,父源和母源染色体上的某些基因不能互相替代。基因由父方传给子女和由母方传递给子女时,常有不同的表现。这是由于基因在生殖细胞(精子和卵子)分化过程中受到不同的修饰。这种现象称为遗传印记(genetic mprinting)。目前认为遗传印记的分子机制主要是DNA的甲基化。已知甲基化可以抑制基因的表达。一些基因在精子和卵子的甲基化状态不同,因此使该基因的表达况有所不同。高度甲基化的被印记的基因可以不表达或表达程度很低,因而可影响到疾病的发生、外显率、表现度,甚至遗传方式。存在这种基因外的修饰现象,也可以解释何以来自父方和母方的某些基因不能互相替代,也可以说明一些孟德尔单基因遗传病却表现出非孟德尔遗传方式。这是又被遗传印记修饰、改变的结果。在遗传病基因研究上,还发现了一类疾病存在有三联序列的高度重复现象。这类疾病有脊髓延髓肌萎缩(Kennedy病,CAG三联体重复)、脆性X综合征(CGG三联体重复)、强直性肌萎缩(GCT三联体重复)。在脆性X综合征的CGG重复序列研究中,发现正常人CGG序列重复6~54次,峰值为29,且各种族间无显著差异。正常男性传递者和女性携带者此序列的重复次数可分别增加到82次及83~90次,这种状态可认为属前突变。女性携带者在传递给下代男性患者而发病时,男性患者的CGG重复次数可高达250~4000次。这种三联体重复序列逐渐增加的突变形式,称为不稳定动态突变。由于这种不稳定的动态突变致使一些疾病发生。除上述所举三种疾病外,1993年3月又报道了Huntington舞蹈病,也是CAG三联体重复,但部位在基因前半部与延髓脊髓肌萎缩在后半部不同。遗传病的研究是当代学术进展最快的领域之一,对其发病基础的知识日益丰富。
一、染色体病
染色体病是由于染色体数目或结构异常而发生的疾病。染色体数目异常比结构异常更为常见。
1.数目性染色体畸变 染色体的数目异常可表现为非整倍体(aneuploid),即其数目并非单倍体(haploid)的整倍数,在数目上出现多或少于整倍体,如45或47条染色体。还可表现为多倍体(polyploid),即染色体数目整倍多于单倍体,如三倍体(triploid)69条,四倍体(tetraploid)92条。常见的非整倍体患者是某对染色体不是2条而出现3条,称为三体综合征(trisomic syndrome)。如果某对染色体缺少1条,则称为单体综合征(monosomic syndrome)。多倍体患者很少见,可见于肿瘤细胞和流产胎儿。
染色体数目异常几乎全是减数分裂不分离(non disjunction)或分裂后期迟延(anaphase lag)的结果。在第一或第二次减数分裂时期,由于两条同源染色体未能分开,而造成子代细胞染色体数目或多或少。
2.结构性染色体畸变 这种畸变是在细胞分裂过程中曾有染色体断裂所致。常见的结构异常有缺失、环状染色体、易位、重复、倒位和等臂染色体(图6-1)。
(1)缺失:指染色体丢失一段。即染色体一处断裂,其无着丝粒的一端常丢失,成为末端缺失;染色体两处断裂,可造成中间段的丢失,为中间缺失。由于遗传基因随染色体断片而丢失,可造成该个体患严重的多发缺陷。
(2)环状染色体:一条染色体的两端发生断裂,两侧末端片段丢失,断端相互连接形成环状染色体。
(3)易位:当两条非同源染色体同时发生断裂时,断落片段由一条染色体移至另一条染色体的断端上,形成易位染色体。易位可以是平衡性的,也可以是不平衡性的。在易位发生过程中,可造成染色体的点缺失,基因断裂损伤或位置效应,由此产生表型异常,此种称为不平衡易位(unbalanced translocation)。没有遗传物质的得失者,表型正常,称为平衡性易位(balanced translocation)。这两种易位很难从常规染色体检查中区分。
(4)重复:是指一条染色体的断片移至同源染色体的相应部位上,造成该同源染色体此段重复。
(5)倒位;是指一条染色体两处断裂,中间的片段倒转180°后,再与两断端连接起来,形成倒位,这种倒位可以发生在两臂间,也可发生在臂内。
(6)等臂染色体:是着丝粒分裂异常所致。正常应是纵裂分开,如果横裂分开,短臂与短臂,或长臂与长臂相接,各形成等臂染色体。
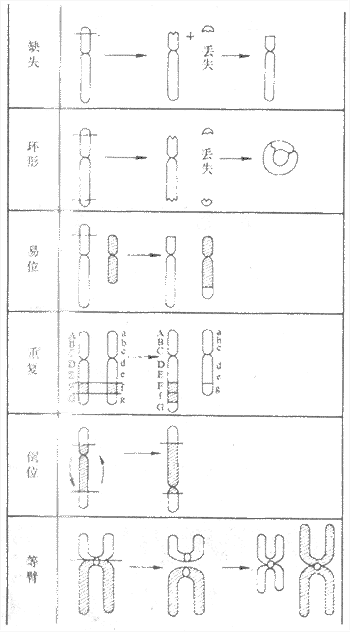
图6-1 人类染色体几种结构畸变的模式图
由上可见,染色体断裂是结构重排、结构异常的重要前提。没有先发生断裂,就没有结构的异常。断裂本是常见的生理现象,减数分裂或有丝分裂中均可发生。用姊妹染色单体交换检查方法可查出。尽管断裂经常发生,但显然大部分断裂可“自愈”。而不引起可查觉的染色体结构变化。因此,断裂后所致染色体的结构畸变的具体原因和机制尚不很清楚。但已了解到有一些因素可引起断裂增加,如射线照射、病毒感染、药物和环境因素等。几种称为染色体断裂综合征(chromosome breakage syndrome)的疾病,如毛细血管扩张性共济失调症(ataxia teleangiectatica)、Bloom综合征、着色性干皮症(xeroderma pigmentosa)和Fanconi贫血等,病人的染色体裂隙和断裂发生率增高。
3.几种染色体病举例
(1)Down综合征:是最常见的常染色体病,其染色体异常在21三体,主要核型为三体型,少数为易位型或嵌合型。表型特征有智力低下、伸舌、鼻梁低平、眼裂上斜、小耳、小颌、枕平、内眦敖皮、颈短及肌张力减低等,常伴有先天性心脏发育缺陷。患者急性淋巴性和粒细胞性白血病的发生率增高。发病率随母龄增长而增高(图6-2)。

图6-2 21三体核型与正常核型比较
上图 21三体(Down)综合征下图 正常核型
(2)Edwards综合征:染色体异常为18三体。表型特征有智力低下、小头、前额窄、枕部突、小颌且张口范围小,腭弓高窄、低位耳、肾畸形(图6-3)、肌张力增高及手紧握等(图6-4)。
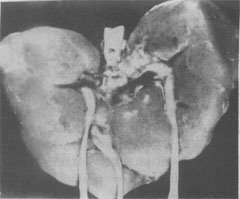
图6-3 18三体(Edwards)综合征的畸形肾
图示马蹄状融合肾及额外输尿管
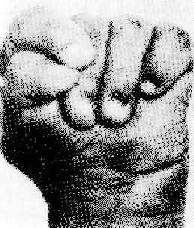
图6-4 (Edwards)综合征的肌张力增高
图示手紧握及特殊拳方式
(3)Patau综合征:染色体异常为13三体。表型特征中有中枢神经系统发育缺陷,呈前脑无裂畸形(holoprosencephaly)(图6-5)。前额小呈斜坡样,头皮后顶部常有缺损。视网膜发育不良,切片镜下观察可见菊形团形成。严重智力低下,唇裂及腭裂、多指等。中性粒细胞核上有过多的突起。
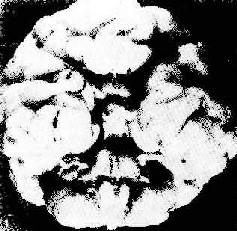
图6-5 13三体(Patau)综合征脑畸形
图示前脑无裂
(4)猫叫综合征(cri du chat syndrome):染色体异常为5号染色体短臂部分或全部缺失。表型特征有:婴儿时期哭声似猫叫,因而得名,可有喉器小和会厌小等喉部发育异常。严重智力低下,小头、圆形脸、眼距宽、眼裂下斜、耳位低、内眦赘皮等。
(5)Turner综合征:这是唯一已知的性染色体单体病(sex chromosome monosomy)。也称先天性卵巢发育不全症。典型核型是45,X。表型特征有:身材矮小,颈蹼及指、趾背部浮肿,为胎儿期淋巴水肿的残迹。后发际低、盾状胸、肘外翻,卵巢发育不全、原发闭经,性器官幼稚型(图6-6)。

图6-6 Turner综合征的一种核型 46,X,r(X)
由于一条X染色体呈环形,末端片段有丢失,影响卵巢发育
(6)Klinefelter综合征:又称先天性睾丸发育不全症。典型的核型是47,XXY。表型特征有睾丸发育不全。身材修长,乃足跟至趾骨间的距离增长所致。男子乳腺发育、阴毛呈女性型分布,阴茎及睾丸均小。严重者伴有智力迟钝、隐睾及尿道下裂等(图6-7)。

图6-7 Klinefelter综合征的核型47,XXY
由于增加一条X染色体,影响睾丸发育
染色体核型分析时,有时可见到一个患者有两种不同的核型。如果这两种不同核型的细胞起源于单一合子(zygote),则这种由两种或两种以上细胞组成的个体称为嵌合体(mosaicism)。起源于一个以上的合子时,称为异源嵌合体(chimera)。如Klinefelter综合征常可见46,XX/47,XXX/48,XXXY等嵌合体。Dwn综合征也可见46,XY(XX)/47,XY(XX),+21的嵌合体。异常核型细胞所占比例少者症状较轻。
二、单基因遗传病
起源于单一(对)基因的突变。单基因遗传病的传递方式是按孟德尔法则(Mendelian law)传至后代的。新突变所致的患者可无家族史。目前已知的单基因遗传病有3000余种。根据突变基因所在的位点和性状的不同,而区分为下列不同类型。
1.常染色体显性遗传病(autosomal dominant disorder)致病基因在常染色体上,等位基因之一突变,杂合状态下即可发病。致病基因可以是生殖细胞发生突变而新产生,也可以是由双亲任何一方遗传而来的。此种患者的子女发病的概率相同,均为1/2。此种患者的异常性状表达程度可不尽相同。在某些情况下,显性基因性状表达极其轻微,甚至临床不能查出,这种情况称为失显(non penetrance)。由于外显不完全,在家系分析时可见到中间一代人未患病的隔代遗传系谱,这种现象又称不规则外显(irreqular dominance)。还有一些常染色体显性遗传病,在病情表现上可有明显的轻重差异,纯合子患者病情严重,杂合子患者病情轻,这种情况称不完全外显(incomplete dominance)。常染色体显性遗传病常见者有Marfan综合征、Ehlers-Danlos综合征、先天性软骨发育不全、多囊肾、结节性硬化、Huntington舞蹈病、家族性高胆固醇血症、神经纤维瘤病、肠息肉病以及视网膜母细胞瘤等。
2.常染色体隐性遗传病(autosomal recessive disorder)致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。子代有1/4的概率患病,子女患病概率均等。许多遗传代谢异常的疾病,属常染色体隐性遗传病。按照“一个基因、一个酶”(one gene one enzyme)或“一个顺反子、一个多肽”(one cistron one polypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性(Wilson病)及半乳糖血症等。
3.性连锁遗传病(sex-linked disorder)以隐性遗传病为多见。致病基因在X染色体上,性状是隐性的,女性大多只是携带者,这类女性携带者与正常男性婚配,子代中的男性有1/2是概率患病,女性不发病,但有1/2的概率是携带者。男性患者与正常女性婚配,子代中男性正常,女性都是携带者。因此X连锁隐性遗传在患病系中常表现为女性携带,男性患病。男性的致病基因只能随着X染色体传给女儿,不能传给儿子,称为交叉遗传。这类常见的疾病有血友病A、假性肥大性肌营养不良症(Duchenne肌营养不良),红绿色盲等。其中红绿色盲如女性携带者和男性患者婚配,子代中的男性有1/2的概率患病,而女性可有1/2的概率患病及1/2概率为携带者。
X连锁显性遗传病病种较少,有抗维生素D性佝偻病等。这类病女性发病率高,这是由于女性有两条X染色体,获得这一显性致病基因的概率高之故,但病情较男性轻。男性患者病情重,他的全部女儿都将患病。
Y连锁遗传病的特点是男性传递给儿子,女性不发病。因Y染色体上主要有男性决定因子方面的基因,其他基因很少,故Y连锁遗传病极少见。
三、多基因遗传病
人类许多生理特征如身高、体重、血压和肤色深浅,毛发疏密等,是受多项基因决定的。多基因遗传病是由两对以上微效基因共同作用造成的,无显性和隐性之分。每对基因作用较小,但有积累效应。在发病时还常受环境因素的影响,故也称多因子遗传(multifactorial inheritance)。这类疾病中遗传因素所起的作用程度不同,按其程度大小以百分率(%)来表示,称为遗传度。环境因素影响越大,遗传度越低。唇裂、腭裂、高血压、糖尿病、躁狂抑郁症、类风湿性关节炎及先天性心脏病等,均有多基因遗传基础,并各自有其遗传度。
要确定某一疾病为多基因遗传病,是比较困难的,首先要除外染色体病和单基因遗传病,还要进行较为周密的家系调查。
作为遗传病的发病基础,近年来的认识越加深入。已往知之较多的是染色体畸变,畸变处出现染色体遗传物质的得失、断裂或位置改变而使机体出现病状。另一方面是基因突变,是编码基因的碱基序列发生变化,随之表达和表达产物异常。这种基因突变引起的疾病,随该基因所在染色体连锁、交换,按孟德尔的自由组合及分离律传递给下一代,称为孟德尔式遗传病。人类线粒体DNA是细胞核外DNA,它的DNA突变也可致线粒体功能异常出现疾病,总称线粒体遗传病。线粒体遗传病是由卵细胞胞浆内突变的线粒体DNA传递给下一代的。因此表现为母系遗传。家系中可见男女均可患病,但男患者后代再无患者。近年来人类基因研究发现,父源和母源染色体上的某些基因不能互相替代。基因由父方传给子女和由母方传递给子女时,常有不同的表现。这是由于基因在生殖细胞(精子和卵子)分化过程中受到不同的修饰。这种现象称为遗传印记(genetic mprinting)。目前认为遗传印记的分子机制主要是DNA的甲基化。已知甲基化可以抑制基因的表达。一些基因在精子和卵子的甲基化状态不同,因此使该基因的表达况有所不同。高度甲基化的被印记的基因可以不表达或表达程度很低,因而可影响到疾病的发生、外显率、表现度,甚至遗传方式。存在这种基因外的修饰现象,也可以解释何以来自父方和母方的某些基因不能互相替代,也可以说明一些孟德尔单基因遗传病却表现出非孟德尔遗传方式。这是又被遗传印记修饰、改变的结果。在遗传病基因研究上,还发现了一类疾病存在有三联序列的高度重复现象。这类疾病有脊髓延髓肌萎缩(Kennedy病,CAG三联体重复)、脆性X综合征(CGG三联体重复)、强直性肌萎缩(GCT三联体重复)。在脆性X综合征的CGG重复序列研究中,发现正常人CGG序列重复6~54次,峰值为29,且各种族间无显著差异。正常男性传递者和女性携带者此序列的重复次数可分别增加到82次及83~90次,这种状态可认为属前突变。女性携带者在传递给下代男性患者而发病时,男性患者的CGG重复次数可高达250~4000次。这种三联体重复序列逐渐增加的突变形式,称为不稳定动态突变。由于这种不稳定的动态突变致使一些疾病发生。除上述所举三种疾病外,1993年3月又报道了Huntington舞蹈病,也是CAG三联体重复,但部位在基因前半部与延髓脊髓肌萎缩在后半部不同。遗传病的研究是当代学术进展最快的领域之一,对其发病基础的知识日益丰富。
一、染色体病
染色体病是由于染色体数目或结构异常而发生的疾病。染色体数目异常比结构异常更为常见。
1.数目性染色体畸变 染色体的数目异常可表现为非整倍体(aneuploid),即其数目并非单倍体(haploid)的整倍数,在数目上出现多或少于整倍体,如45或47条染色体。还可表现为多倍体(polyploid),即染色体数目整倍多于单倍体,如三倍体(triploid)69条,四倍体(tetraploid)92条。常见的非整倍体患者是某对染色体不是2条而出现3条,称为三体综合征(trisomic syndrome)。如果某对染色体缺少1条,则称为单体综合征(monosomic syndrome)。多倍体患者很少见,可见于肿瘤细胞和流产胎儿。
染色体数目异常几乎全是减数分裂不分离(non disjunction)或分裂后期迟延(anaphase lag)的结果。在第一或第二次减数分裂时期,由于两条同源染色体未能分开,而造成子代细胞染色体数目或多或少。
2.结构性染色体畸变 这种畸变是在细胞分裂过程中曾有染色体断裂所致。常见的结构异常有缺失、环状染色体、易位、重复、倒位和等臂染色体(图6-1)。
(1)缺失:指染色体丢失一段。即染色体一处断裂,其无着丝粒的一端常丢失,成为末端缺失;染色体两处断裂,可造成中间段的丢失,为中间缺失。由于遗传基因随染色体断片而丢失,可造成该个体患严重的多发缺陷。
(2)环状染色体:一条染色体的两端发生断裂,两侧末端片段丢失,断端相互连接形成环状染色体。
(3)易位:当两条非同源染色体同时发生断裂时,断落片段由一条染色体移至另一条染色体的断端上,形成易位染色体。易位可以是平衡性的,也可以是不平衡性的。在易位发生过程中,可造成染色体的点缺失,基因断裂损伤或位置效应,由此产生表型异常,此种称为不平衡易位(unbalanced translocation)。没有遗传物质的得失者,表型正常,称为平衡性易位(balanced translocation)。这两种易位很难从常规染色体检查中区分。
(4)重复:是指一条染色体的断片移至同源染色体的相应部位上,造成该同源染色体此段重复。
(5)倒位;是指一条染色体两处断裂,中间的片段倒转180°后,再与两断端连接起来,形成倒位,这种倒位可以发生在两臂间,也可发生在臂内。
(6)等臂染色体:是着丝粒分裂异常所致。正常应是纵裂分开,如果横裂分开,短臂与短臂,或长臂与长臂相接,各形成等臂染色体。
图6-1 人类染色体几种结构畸变的模式图
由上可见,染色体断裂是结构重排、结构异常的重要前提。没有先发生断裂,就没有结构的异常。断裂本是常见的生理现象,减数分裂或有丝分裂中均可发生。用姊妹染色单体交换检查方法可查出。尽管断裂经常发生,但显然大部分断裂可“自愈”。而不引起可查觉的染色体结构变化。因此,断裂后所致染色体的结构畸变的具体原因和机制尚不很清楚。但已了解到有一些因素可引起断裂增加,如射线照射、病毒感染、药物和环境因素等。几种称为染色体断裂综合征(chromosome breakage syndrome)的疾病,如毛细血管扩张性共济失调症(ataxia teleangiectatica)、Bloom综合征、着色性干皮症(xeroderma pigmentosa)和Fanconi贫血等,病人的染色体裂隙和断裂发生率增高。
3.几种染色体病举例
(1)Down综合征:是最常见的常染色体病,其染色体异常在21三体,主要核型为三体型,少数为易位型或嵌合型。表型特征有智力低下、伸舌、鼻梁低平、眼裂上斜、小耳、小颌、枕平、内眦敖皮、颈短及肌张力减低等,常伴有先天性心脏发育缺陷。患者急性淋巴性和粒细胞性白血病的发生率增高。发病率随母龄增长而增高(图6-2)。
图6-2 21三体核型与正常核型比较
上图 21三体(Down)综合征下图 正常核型
(2)Edwards综合征:染色体异常为18三体。表型特征有智力低下、小头、前额窄、枕部突、小颌且张口范围小,腭弓高窄、低位耳、肾畸形(图6-3)、肌张力增高及手紧握等(图6-4)。
图6-3 18三体(Edwards)综合征的畸形肾
图示马蹄状融合肾及额外输尿管
图6-4 (Edwards)综合征的肌张力增高
图示手紧握及特殊拳方式
(3)Patau综合征:染色体异常为13三体。表型特征中有中枢神经系统发育缺陷,呈前脑无裂畸形(holoprosencephaly)(图6-5)。前额小呈斜坡样,头皮后顶部常有缺损。视网膜发育不良,切片镜下观察可见菊形团形成。严重智力低下,唇裂及腭裂、多指等。中性粒细胞核上有过多的突起。
图6-5 13三体(Patau)综合征脑畸形
图示前脑无裂
(4)猫叫综合征(cri du chat syndrome):染色体异常为5号染色体短臂部分或全部缺失。表型特征有:婴儿时期哭声似猫叫,因而得名,可有喉器小和会厌小等喉部发育异常。严重智力低下,小头、圆形脸、眼距宽、眼裂下斜、耳位低、内眦赘皮等。
(5)Turner综合征:这是唯一已知的性染色体单体病(sex chromosome monosomy)。也称先天性卵巢发育不全症。典型核型是45,X。表型特征有:身材矮小,颈蹼及指、趾背部浮肿,为胎儿期淋巴水肿的残迹。后发际低、盾状胸、肘外翻,卵巢发育不全、原发闭经,性器官幼稚型(图6-6)。
图6-6 Turner综合征的一种核型 46,X,r(X)
由于一条X染色体呈环形,末端片段有丢失,影响卵巢发育
(6)Klinefelter综合征:又称先天性睾丸发育不全症。典型的核型是47,XXY。表型特征有睾丸发育不全。身材修长,乃足跟至趾骨间的距离增长所致。男子乳腺发育、阴毛呈女性型分布,阴茎及睾丸均小。严重者伴有智力迟钝、隐睾及尿道下裂等(图6-7)。
图6-7 Klinefelter综合征的核型47,XXY
由于增加一条X染色体,影响睾丸发育
染色体核型分析时,有时可见到一个患者有两种不同的核型。如果这两种不同核型的细胞起源于单一合子(zygote),则这种由两种或两种以上细胞组成的个体称为嵌合体(mosaicism)。起源于一个以上的合子时,称为异源嵌合体(chimera)。如Klinefelter综合征常可见46,XX/47,XXX/48,XXXY等嵌合体。Dwn综合征也可见46,XY(XX)/47,XY(XX),+21的嵌合体。异常核型细胞所占比例少者症状较轻。
二、单基因遗传病
起源于单一(对)基因的突变。单基因遗传病的传递方式是按孟德尔法则(Mendelian law)传至后代的。新突变所致的患者可无家族史。目前已知的单基因遗传病有3000余种。根据突变基因所在的位点和性状的不同,而区分为下列不同类型。
1.常染色体显性遗传病(autosomal dominant disorder)致病基因在常染色体上,等位基因之一突变,杂合状态下即可发病。致病基因可以是生殖细胞发生突变而新产生,也可以是由双亲任何一方遗传而来的。此种患者的子女发病的概率相同,均为1/2。此种患者的异常性状表达程度可不尽相同。在某些情况下,显性基因性状表达极其轻微,甚至临床不能查出,这种情况称为失显(non penetrance)。由于外显不完全,在家系分析时可见到中间一代人未患病的隔代遗传系谱,这种现象又称不规则外显(irreqular dominance)。还有一些常染色体显性遗传病,在病情表现上可有明显的轻重差异,纯合子患者病情严重,杂合子患者病情轻,这种情况称不完全外显(incomplete dominance)。常染色体显性遗传病常见者有Marfan综合征、Ehlers-Danlos综合征、先天性软骨发育不全、多囊肾、结节性硬化、Huntington舞蹈病、家族性高胆固醇血症、神经纤维瘤病、肠息肉病以及视网膜母细胞瘤等。
2.常染色体隐性遗传病(autosomal recessive disorder)致病基因在常染色体上,基因性状是隐性的,即只有纯合子时才显示病状。此种遗传病父母双方均为致病基因携带者,故多见于近亲婚配者的子女。子代有1/4的概率患病,子女患病概率均等。许多遗传代谢异常的疾病,属常染色体隐性遗传病。按照“一个基因、一个酶”(one gene one enzyme)或“一个顺反子、一个多肽”(one cistron one polypeptide)的概念,这些遗传代谢病的酶或蛋白分子的异常,来自各自编码基因的异常。常见的常染色体隐性遗传病有溶酶体贮积症,如糖原贮积症、脂质贮积症、粘多糖贮积症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆状核变性(Wilson病)及半乳糖血症等。
3.性连锁遗传病(sex-linked disorder)以隐性遗传病为多见。致病基因在X染色体上,性状是隐性的,女性大多只是携带者,这类女性携带者与正常男性婚配,子代中的男性有1/2是概率患病,女性不发病,但有1/2的概率是携带者。男性患者与正常女性婚配,子代中男性正常,女性都是携带者。因此X连锁隐性遗传在患病系中常表现为女性携带,男性患病。男性的致病基因只能随着X染色体传给女儿,不能传给儿子,称为交叉遗传。这类常见的疾病有血友病A、假性肥大性肌营养不良症(Duchenne肌营养不良),红绿色盲等。其中红绿色盲如女性携带者和男性患者婚配,子代中的男性有1/2的概率患病,而女性可有1/2的概率患病及1/2概率为携带者。
X连锁显性遗传病病种较少,有抗维生素D性佝偻病等。这类病女性发病率高,这是由于女性有两条X染色体,获得这一显性致病基因的概率高之故,但病情较男性轻。男性患者病情重,他的全部女儿都将患病。
Y连锁遗传病的特点是男性传递给儿子,女性不发病。因Y染色体上主要有男性决定因子方面的基因,其他基因很少,故Y连锁遗传病极少见。
三、多基因遗传病
人类许多生理特征如身高、体重、血压和肤色深浅,毛发疏密等,是受多项基因决定的。多基因遗传病是由两对以上微效基因共同作用造成的,无显性和隐性之分。每对基因作用较小,但有积累效应。在发病时还常受环境因素的影响,故也称多因子遗传(multifactorial inheritance)。这类疾病中遗传因素所起的作用程度不同,按其程度大小以百分率(%)来表示,称为遗传度。环境因素影响越大,遗传度越低。唇裂、腭裂、高血压、糖尿病、躁狂抑郁症、类风湿性关节炎及先天性心脏病等,均有多基因遗传基础,并各自有其遗传度。
要确定某一疾病为多基因遗传病,是比较困难的,首先要除外染色体病和单基因遗传病,还要进行较为周密的家系调查。
相关热词搜索:










论坛新帖
医学推广
频道本月排行
- 8细胞和组织的适应、损伤与修复
- 5再生
- 5冠状动脉性心脏病
- 4神经系统对损伤的基本反应
- 3血栓形成
- 3动脉炎
- 2感染性疾病
- 2食管疾病
- 2胰腺疾病
- 2滋养层细胞肿瘤
热门购物
评论排行
- 2011年临床执业医师考试实践技能真...(13)
- 腋臭手术视频(11)
- 2008年考研英语真题及参考答案(5)
- 节食挑食最伤女人的免疫系统(5)
- 核辐射的定义和单位(5)
- CKD患者Tm与IMT相关(5)
- 齐鲁医院普外科开展“喉返神经监护...(5)
- windows7激活工具WIN7 Activation v1.7(5)
- 正常微循环(5)
- 美大学性教育课来真的 男女上阵亲...(4)