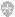缺血/再灌注损伤的机制
2011-12-02 09:16:58 来源: 作者: 评论:0 点击:
关于再灌注损伤的发生机制问题,到目前为止,学说虽多而且有的学说也颇为有力,便尚未得到彻底阐明。兹介绍如下:
一、无复流现象
无复流现象(no-reflow phenomenon)是在犬的实验中发现的。结扎犬的冠状动脉造成局部心肌缺血后,再打开结扎的动脉,使血流重新开放,缺血区并不能得到充分的灌注,故称此现象为无复流或无再灌。这种无复流现象不仅见于心肌,而且也见于脑、肾骼肌缺血后再灌注时。即再灌注损伤实际上是缺血的延续和叠加,缺血细胞并未能得到血液灌注,而是继续缺血,因而损伤加重。所以发生无复流现象,可能与下列因素有关(以心肌为例):
(一)心肌细胞肿胀 由于缺血引起细胞膜Na+-K+泵功能障碍,从而使钠、水在细胞内潴留,因而再灌注时缺血区心肌细胞发生肿胀,压迫微血管。
(二)血管内皮细胞肿胀 缺血及再灌注时也发生内皮细胞肿胀,内皮细胞向管腔伸出突起造成管腔狭窄,阻碍血液灌流。内皮细胞的肿胀与氧自由基的增多有关,因为氧自由基可以使血管内皮细胞膜受损,水钠乃进入内皮细胞而引起细胞水肿。
(三)心肌细胞的收缩缺血所致的心肌细胞收缩形成严重收缩带,压迫微血管,使缺血区某部分得不到血液重新灌注。心肌细胞的肿胀与收缩带可同时存在。
(四)微血管堵塞 Feinburg及其同事曾证明,缺血一定时间后血管内血小板的沉积增加2倍。又证明心肌及肠管缺血后的无复流区内白细胞(主要是中性粒细胞)的聚集明显增加,从组织学上可见白细胞嵌顿、阻塞毛细血管。正常灌注情况下,每2433μm到3261μm长的毛细血管可发现一个白细胞,而在缺血时增加10倍,平均292μm长的毛细血管即有一个白细胞。缺血时红细胞作叠连状聚集,但这不是血管阻塞的主要原因。因为叠连状红细胞的解聚较白细胞与内皮细胞粘着的分离要容易得多。此外也有人解释无复流现象是由于纤维蛋白塞和微血栓形成所致。但有人在再灌前用链激酶进行纤溶并未减轻无复流现象。
微血管的堵塞还与花生四烯酸的代谢产物前列环毒(PGI2)和血栓素A2(TXA2)之间的失衡密切相关。PCI2主要由血管内皮生成,除了有很强的扩血管作用以外,还能抑制血小板的聚集。TXA2主要由血小板生成,不仅是很强的缩血管物质,而且也是一种引起血小板聚集的因子,因此是一个很强的致血栓形成的物质。缺血缺氧时,一方面因为血管内皮细胞受损而致PCI2生成减少,另方面缺氧又可使血小板释放TXA2增多,因而发生强烈的血管收缩和血小板的聚集并进一步释放TXA2,从而促使血栓形成和血管堵塞。动物实验也证明,应用TXA2合成酶抑制药可以使缺血/再灌注以后的冠脉血流改善。
二、钙超载
如前所述,钙反常(无钙灌流后用含钙溶液再灌注)时细胞内钙超载,引起严重的功能及结构障碍。钙反常的原因不甚明了,但主要的损伤是在无钙灌流期出现的细胞膜外板(external lamina)与糖被膜(glycocalyx)表面的分离(两者由Ca2+连结在一起)。细胞膜的这种损伤为再灌注时钙的大量内流提供了条件。在长期缺血缺氧后再给氧或再灌注时也可引起细胞内钙超载。
缺血后再灌注时细胞内钙超载的机制尚无定论,但可能与下列因素有关,如图11-1哺乳类钙代谢模式图所示:

图11-1 哺乳类细胞钙代谢模式图
①电压依赖性钙通道;②质膜钙泵ATP酶;③Na+-Ca2+交换;④线粒体;⑤肌浆网;⑥细胞内蛋白或阴离子结合的钙;⑦膜磷脂的极性头部;⑧结合于质膜糖被的钙
(一)钠的平衡障碍 有人证明,再灌注时钙超载是由于钠平衡障碍所致。因为在缺血缺氧时发生了细胞酸中毒,细胞内pH值降低,所以在再灌注时细胞内外形成pH梯度差,由于Na+-H+交换,致细胞内钠增加,然后又依Na+-Ca2+交换机制使细胞外钙大量内流造成细胞钙超载。
(二)细胞膜通透性增高缺血缺氧引起的细胞酸中毒在再灌注时通过细胞内外Na+-H+交换和Na+-Ca2+交换而使细胞内钙增加,而细胞内钙增加可激活磷脂酶,使膜磷脂降解,细胞膜通透性增高,故在灌注时细胞外钙顺着浓度梯度而大量内流,细胞膜通透性增高的更重要的原因可能是再灌注时氧自由基的大量产生。氧自由基可引发细胞膜的脂质过氧化,使膜受损,通透性增高。
(三)线粒体受损 有些学者认为原发性损伤在于线粒体。如所周知,缺血时线粒体结构的功能障碍出现最早,表现为线粒体肿胀、嵴断裂。线粒体膜流动性降低,氧化磷酸化功能受损ATP生成障碍。因此,使上述损伤更为严重。
ATP减少使肌膜及肌浆网膜钙泵功能障碍,由于钙泵功能障碍不能排出和摄取细胞浆中过多的钙,致使细胞浆中游离钙浓度增加而造成钙超载。细胞浆中过多的钙最终形成磷酸盐沉积于线粒体,使线粒体结构及功能更加破坏。
细胞钙超载是再灌注损伤的一个重要特征,但目前,仍未能搞清它究竟是再灌注损伤的原因抑制或结果。
三、白细胞的作用
白细胞(主要是中性粒细胞)出现于梗塞的心肌中已为尸检所证实。1984年Mullane及其同事证明,冠状动脉堵塞60分钟时心肌组织就有白细胞出现,5小时后在缺血区有大量的白细胞聚集。根据Engler及其同事的研究,再灌注时白细胞数非但不减少反而增加。以犬心肌缺血为模型,再灌注仅5分钟,心内膜中性粒细胞就增加25%,缺血轻的组织白细胞聚集也少。
组织缺血和再灌注时白细胞浸润增加的机制还不十分清楚。可能是由于组织受损时,细胞膜磷脂降解,花生四烯酸代谢产物增多,其中有些物质具有很强的趋化作用,因而就能吸引大量白细胞进入组织或粘附于血管内皮,而白细胞本身又能释放很多具有趋化作用的炎性介质,如白三烯之一的LTB4,从而使微循环中白细胞进一步增加。
白细胞积聚对组织的损伤作用在于:
(一)嵌顿、堵塞毛细血管有助于形成无复流现象。微动脉及微静脉亦有大量白细胞粘附于内皮细胞,虽不一定堵塞血流,但粘附的白细胞仍可损伤组织并释放趋化因子从而吸引更多的细胞。
(二)白细胞可以增加血管通透性,水肿组织的含水量与白细胞密度呈正相关,说明白细胞可能引发水肿。白细胞增加血管通透性、引发水肿的机制与白细胞释放的某些炎症介质有关。
(三)激活的中性粒细胞释放溶酶体酶,可使组织发生蛋白水解性破坏和液化。
(四)中性粒细胞可通过产生氧自由基而损伤组织。
白细胞在缺血再灌注损伤的作用,可被以下实验结果证明:
1.用除去白细胞的血液进行再灌注,可以防止水肿产生并减轻再灌性损伤。
2.抗炎药减轻组织白细胞浸泣,可缩小梗塞面积。实验证明布洛芬(ibuprofen)对心肌具有保护作用,主要是由于抑制白细胞浸润的作用所致。
3.用补体抑制药降低补体,从而减少白细胞浸润,可能减轻组织损伤。丝氨酸蛋白酶抑制药,可以缩小心肌梗塞,而这种酶恰是一种在心肌缺血时能激活补体的酶。因此这种酶的抑制药,可通过抑制补体的激活而抑制白细胞浸润,从而减轻心肌损伤。
四、高能磷酸化合物的缺乏
心肌正常情况下以有氧代谢形式生成三磷酸腺苷(ATP)供作功需要。心肌缺血时则转为无氧代谢为主,ATP合成减少,以致心舒缩功能障碍。在犬的实验中证明,心肌严重缺血15分钟(结扎冠状动脉左旋支),心肌发生可逆性损伤。此时如果得到血液再灌注,则细胞并不死亡。但有很多报告指出,短时间缺血后,收缩功能长时间不能恢复。究其原因,多认为与ATP水平的低下有关。研究证明缺血15分钟时不仅ATP减少60%。总腺苷酸池也减少50%。ADP也轻度减少(可能转为ATP或AMP),AMP明显升高,但其升高程度小于ATP减少辐度。再灌注20分钟ATP明显回升,但只接近正常的一半,再灌注24小时仍然维持在低水平上,只有在再灌注4天后ATP及总腺苷池才近于恢复,但仍低于非缺血区(图11-2)。

图11-2 心肌缺血/再灌注时ATP、Ca2+、K+的变化
以大鼠离体作功心脏为模型(Neely氏模型),先在20℃低温下给心脏停跳液,再短时间全心缺血(夹住主动脉)后,再灌注生理溶液。结果是给停跳液30分钟再缺血30分钟后,ATP几乎完全丧失,ADP明显减少,AMP明显增加,总腺苷酸量显著降低。再灌注60分钟可使ATP明显回升,但不及正常对照的一半,而总腺苷酸量则明显低于给停跳液后,比正常对照减少50%。上述研究提示缺血及再灌注损伤的心肌有氧代谢性发生严重损伤,影响能量代谢及心肌功能的恢复。
再灌注时高能磷酸化合物之所以恢复慢且总腺苷酸水平明显下降,可能与下列因素有关。
(一)缺血心肌的代谢障碍主要表现为对氧的利用能力受限,有氧代谢严重受损。在缺血进入不可逆阶段再灌注时,氧的利用并不增加,心肌只能利用运至心肌的氧的17%。氧的利用能力受限与缺血及再灌注所致线粒体受损有关。
(二)ATP合成的前身物质(腺苷、肌苷、次黄嘌呤等)在再灌时被冲洗出去,使心肌失去再合成高能磷酸化合物的物质基础。实验证明在再灌注液中补充肌苷或谷氨酸能促进ATP的合成及心功能的恢复。
3.线粒体膜发生氧自由基诱发的脂质过氧化反应使线体受损。线粒体膜富有磷脂,线粒体在缺氧时又是产生自由基的场所,因此极易引起膜脂过氧化使线粒体功能障碍。
五、自由基的作用
自由基(free radical)是具有一个不配对电子的原子和原子团的总称。由氧诱发的自由基称为氧自由基或活性氧,如超氧阴离子(O2)、羟自由基(OH.)及单线态氧(1O2,激发态放出一个光子)等非脂性自由基。H2O2非自身基,但也是一种氧化作用很强的活性氧。氧自由基与多聚不饱和脂肪酸作用后生成的中间代谢产物烷自由基(L.)、烷氧基(LO.)、烷过氧基(LOO.)等属于脂性自由基。氧自由基和脂性自由基的性质极为活泼,易于失去电子(氧化)或夺取电子(还原),特别是其氧化作用强,故具有强烈的引发脂质过氧化的作用。在生理情况下,氧通常是通过细胞色素氧化酶系统接受4个电子还原成水,同时释放能量,但也有1~2%的氧接受一个电子生成O2,或再接受一个电子生成H2O2。但由于细胞内存有超氧化物歧化酶(superoxide dismutase, SOD)和谷胱甘肽过氧化物酶(glrtathione peroxidase, GSH-PX)等抗氧化酶类可以及时清除它们,所以对机体并无有害影响。在病理条件下,由于活性氧产生的过多或抗氧化酶类活性下降,则可引发链式脂质过氧化反应损伤细胞膜系并进而使细胞死亡。
(一)细胞内氧自由基的生成
分子氧在线粒体细胞色素氧化酶系统中接受一个电子而被还原生成O2。
O2e- → O2-
这是其他活性氧产生的基础。过氧化氢(H2O2)及羟自由基(OH.)续发于此。即氧在获得一个电子时还原生成O2-,获得2个电子生成H2O2,获得3个电子生成OH.,获得四个电子生成H2O。

H2O2既可由O2-自发歧化产生,也可经酶促歧化而生成。
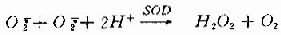
H2O2本身并非自由基而是一种活性氧,它与氧自由基的产生有密切关系(详下文)。
OH.自由基的产生不仅需要O2-或H2O2,而且要有过渡金属,如铁的螯合物的存在。由铁催化的Fenton型Haber-Weiss反应可迅速形成OH.,而单纯的Haber-W-eiss反应速度很慢,很难由此形成OH.。
O2-+H2O2→O2+OH-+OH.
(Haber-Weiss反应)
Fe(Ⅲ)+O2-→fe(Ⅱ)+O2 (2)
Fe(Ⅱ)H2O2 →Fe(Ⅲ)+OH-+OH. (3)
也就是说,O2-使铁还原,还原的铁再使H2O2还原生成OH.,OH.是最活跃最强力的氧自由基。缺血与再灌注时氧自由基生成过多,其机制可能是:
1.黄嘌呤氧化酶的形成增多 黄嘌呤氧化酶(xanthine ocidase, XO)的前身是黄嘌呤脱氢酶(xanthine dehydrogenase, XD)。这两种酶主要存在于毛细血管内皮细胞内。正常时只有10%以XO的形式存在,90%为XD。缺血时由于ATP减少,膜泵功能失灵,Ca2+进入细胞激活Ca2+依赖性蛋白水解酶,使XD大量转变为XO。缺血时ATP不能用来释放能量,而且还依次降解为ADP、AMP和次黄嘌呤,故在缺血组织内次黄嘌呤大量堆积。再灌注时,大量分子氧随血液进入缺血组织,黄嘌呤氧化酶在催化次黄嘌呤转变为黄嘌呤并进而催化黄嘌吟转变为尿酸的两步反应中,都同时以分子氧为电子接受体,从而产生大量的O2-和H2O2,后者再在金属离子参与下形成OH.。因此,再灌注时组织内O2-、OH.等氧自由基大量增加。
2.中性粒细胞 中性粒细胞在吞噬活动时耗氧量显著增加,所摄取的O2绝大部分经细胞内的NADPH氧化酶和NADH氧化酶的作用而形成氧自由基,并用以杀灭病原微生物。如氧自由基产生过多或机体清除氧自由基的酶系统活性不足或抗氧化剂不够时,中性粒细胞形成的氧自由基就可损害组织。
在再灌注时,由黄嘌呤氧化酶的作用所产生的氧自由基起原发的、主要的作用;这些自由基作用于细胞膜后产生的具有趋化活性的物质如LTB4等可吸引大量中性粒细胞到局部释放氧自由基等物质而进一步损害组织。
3.线粒体 可能是由于缺氧使ATP减少,Ca2+进入线粒体增多而使线粒体功能受损,细胞色素氧化酶系统功能失调,以致进入细胞内的氧,经单电子还原而形成的氧自由基增多而经4价还原而形成的水减少。细胞色素氧化酶的功能失调,也可能是缺氧时细胞内氧分压降低的结果。
4.儿茶酚胺的增加 交感-肾上腺髓质系统是机体在应激时的重要调节系统。在各种应激包括缺氧的条件下,此系统分泌大量的儿茶酚胺,儿茶酚胺一方面具有重要的代偿调节作用,但过多的儿茶酚胺特别是它的氧化产物,往往又成为对机体的有害因素。实验证明,大量的异丙肾上腺素、去甲肾上腺素、肾上腺素均能引起细胞损伤。造成心肌损害的是儿茶酚胺的氧化产物,而非儿茶酚胺本身。儿茶酚胺氧化能产生具有细胞毒性的氧自由基。肾上腺素代谢产生紧上腺素红的过程中有O2-产生。
(二)自由基反应与再灌注损伤
机体在生命过程中所能遇到的自由基种类很多。因此很难概括其生物学反应。自由基参与一个反应系统后能形成新的自由基。因此自由基一旦形成,就成为自由基反应扩展程序的一部分,例如:
R.+XH→RH+H.
R.+CCl4→RCl+Cl3C.
另一个自由基反应则是自由基加入到不饱和键中去。如脂肪酸及芳香族环的不饱和键。
自由基反应既可经自由基中间代谢产物不断向前发展,又可由细胞损伤而终止。自由基反应的扩展可以是无限的,但又可为各种自由基清除剂(free radical scavenger)所终止。
由于自由基有极为活泼的反应性,所以它们能和各种细胞成分(膜磷脂、蛋白、核酸)发生反应。
(1)膜脂:是构成膜脂质双层的重要结构及功能成分,富含不饱和脂肪酸,自由基与不饱和脂肪酸作用引发脂质过氧化(lipid peroxidation)反应。脂质过氧化物的形成使膜受体、膜蛋白酶和离子通道的脂质微环境改变,从而改变它们功能,由于脂质过氧化反应的增强,细胞膜内多价不饱和脂肪酸减少,生物膜不饱和脂肪酸/蛋白质比例失常,膜的液态性、流动性改变,通透性增强。含双键脂肪酸过氧化可生成丙二醛,它的产生与脂质过氧化相平行,因而测定丙二醛含量可代表脂质过氧化物的浓度。丙二醛能使膜成分之间形成交联和聚合(polymerization),使膜的基本特性如变构、离子传递、酶活性等发生改变(图11-3)。
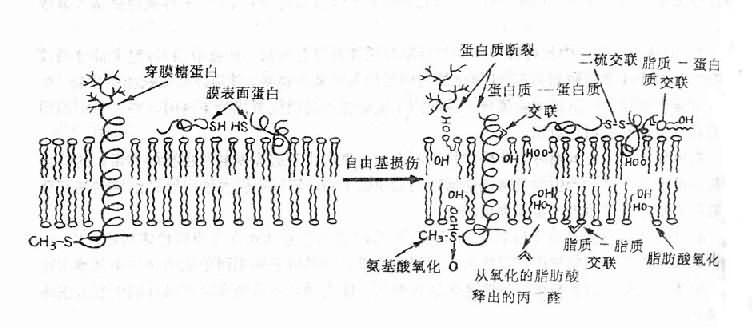
图11-3 自由基对膜的损伤
自由基可通过诱致过氧化而影响脂质,从而产生短链脂酰衍化物和副产物
丙二醛。丙二醛反应可介导各种交联反应。自由基也能催化氨基
酸氧化、蛋白质-蛋白质交联和蛋白质链的断裂
(2)蛋白质:在自由基的作用下,胞浆及膜蛋白及某些酶可交联成二聚体或更大的聚合物。这种交联既可借助于蛋白质之间的二硫键形成也可由于自由基损伤的氨基酸残基间的反应形成。蛋白质的交联将使其失去活性,结构改变。
(3)核酸:自由基对细胞的毒性作用主要表现为染色体畸变,核酸碱基改变或DNA断裂。80%是OH.的作用。OH.易与脱氧核糖及硷基反应并使其改变。
(三)细胞对自由基损伤的防护
自由基的产生既然是有机体在正常或病理条件下的常见现象,因此在进化过程中也就形成了一系列对抗自由基,防止其损伤的系统。这种生化学防护系统主要有两大类:低分子自由基清除剂及复合酶系统。
1.低分子清除剂 存在于细胞脂质部分的自由基清除剂有维生素E(α-生育酚)和维生素A(β-胡萝卜素);存在于细胞内外水相中的自由基清除剂有半胱氨酸、抗坏血酸和谷胱甘肽等,它们能提供电子使自由基还原,故有重要的防护作用。例如维生素E(生育酚)能还原O2-、单线态氧(1O2)、过氧化脂质自由基等;抗坏血酸具有相同作用而且能协助生育酚维持其具有活性的还原状态。β-胡萝卜素是单线态氧(1O2)的有效清除剂并能抑制脂质过氧化。
胞浆中的还原型谷胱甘肽(GSH)与还原型辅酶Ⅱ(NADPH)在某些酶如过氧化氢酶、谷胱甘肽过氧化物酶等的协同作用下,能还原H2O2、过氧化脂质、二硫化物及某些自由基。
2.酶性清除剂 细胞内有二种酶可以清除H2O2,即过氧化氢酶和过氧化物酶。如所周知H2O2是OH.自由基的前身,上述两酶可使H2O2浓度降低,从而避免高毒性OH.的产生。这两个酶的细胞内分布,尚不清楚。
细胞内具有清除剂作用的另一个重要酶是超氧化物歧化酶(SOD)。它是一种金属蛋白,可以歧化O2-生成H2O2。哺乳类细胞含有两种SOD。其一是位于胞浆中的CuZn 超氧化物歧化酶,另一是位于线粒体中的Mn超氧化物歧化酶。SOD作用的重要意义,在于清除H2O2及OH.的前身O2-,从而保护细胞不受强毒性氧自由基的损伤。
清除剂的浓度及活性下降必将引起自由基所致之细胞损伤。
(四)自由基在缺血再灌注损伤机制中的地位
缺血/再灌注损伤机制中的各种学说,无一不与自由基的作用有关。
1.缺血/再灌注时脂质过氧化增强(自由基引发),组织及血浆中脂质过氧化物显著增高,超微结构严重受损。给予抗氧化剂如:维生素E、硒(谷胱甘肽过氧化物酶辅基所含金属)及SOD能显著减轻缺血/再灌注损伤。
2.细胞膜脂质过氧化改变膜酶、离子通道的脂质微环境,从而使膜通透性增高,细胞外钙离子内流。膜上Na+-K+-ATP酶失活,可使细胞内Na+升高,Na+-Ca2+交换增强。而使细胞内钙超载。
3.线粒体膜富有磷脂,缺血/再灌注时自由基引发的线粒体膜脂质过氧化或细胞内形成脂质过氧化物作用于线粒体膜,使膜的液态及流动性改变,从而导致线粒体功能障碍,高能磷酸化物产生减少,自由基产生增多。细胞丧失能量贮备。依靠能量的质膜及肌浆网膜钙泵,由于能量不足不能将肌浆中过多的Ca2+泵出或吸收入肌浆网,致使肌细胞内Ca2+浓度增加,加上由细胞外来的Ca2+终于造成细胞内Ca2+超载,成为细胞致死原因。
4.自由基引发的脂质过氧化造成细胞成分间的交联(脂质-脂质交联、蛋白-蛋白交联、脂质-蛋白交联、蛋白-胶原交联),使整个细胞丧失功能。
5.缺血/再灌注时,微粒体及质膜上的脂加氧酶(lipoxygenase)及环加氧酶(cyclooxygenase)激活,催化花生四烯酸代谢,在加强自由基产生及脂质过氧化的同时形成具有高度生物活性的物质,如前列腺素、血栓素等。很多实验证明,缺血特别是再灌注时血栓素形成增加,前列环素形成减少,因而造成微循环障碍,出现无复流现象。
总之,自由基即使不是缺血/再灌注损伤的唯一发病学因素,至少也是甚为重要的环节。
一、无复流现象
无复流现象(no-reflow phenomenon)是在犬的实验中发现的。结扎犬的冠状动脉造成局部心肌缺血后,再打开结扎的动脉,使血流重新开放,缺血区并不能得到充分的灌注,故称此现象为无复流或无再灌。这种无复流现象不仅见于心肌,而且也见于脑、肾骼肌缺血后再灌注时。即再灌注损伤实际上是缺血的延续和叠加,缺血细胞并未能得到血液灌注,而是继续缺血,因而损伤加重。所以发生无复流现象,可能与下列因素有关(以心肌为例):
(一)心肌细胞肿胀 由于缺血引起细胞膜Na+-K+泵功能障碍,从而使钠、水在细胞内潴留,因而再灌注时缺血区心肌细胞发生肿胀,压迫微血管。
(二)血管内皮细胞肿胀 缺血及再灌注时也发生内皮细胞肿胀,内皮细胞向管腔伸出突起造成管腔狭窄,阻碍血液灌流。内皮细胞的肿胀与氧自由基的增多有关,因为氧自由基可以使血管内皮细胞膜受损,水钠乃进入内皮细胞而引起细胞水肿。
(三)心肌细胞的收缩缺血所致的心肌细胞收缩形成严重收缩带,压迫微血管,使缺血区某部分得不到血液重新灌注。心肌细胞的肿胀与收缩带可同时存在。
(四)微血管堵塞 Feinburg及其同事曾证明,缺血一定时间后血管内血小板的沉积增加2倍。又证明心肌及肠管缺血后的无复流区内白细胞(主要是中性粒细胞)的聚集明显增加,从组织学上可见白细胞嵌顿、阻塞毛细血管。正常灌注情况下,每2433μm到3261μm长的毛细血管可发现一个白细胞,而在缺血时增加10倍,平均292μm长的毛细血管即有一个白细胞。缺血时红细胞作叠连状聚集,但这不是血管阻塞的主要原因。因为叠连状红细胞的解聚较白细胞与内皮细胞粘着的分离要容易得多。此外也有人解释无复流现象是由于纤维蛋白塞和微血栓形成所致。但有人在再灌前用链激酶进行纤溶并未减轻无复流现象。
微血管的堵塞还与花生四烯酸的代谢产物前列环毒(PGI2)和血栓素A2(TXA2)之间的失衡密切相关。PCI2主要由血管内皮生成,除了有很强的扩血管作用以外,还能抑制血小板的聚集。TXA2主要由血小板生成,不仅是很强的缩血管物质,而且也是一种引起血小板聚集的因子,因此是一个很强的致血栓形成的物质。缺血缺氧时,一方面因为血管内皮细胞受损而致PCI2生成减少,另方面缺氧又可使血小板释放TXA2增多,因而发生强烈的血管收缩和血小板的聚集并进一步释放TXA2,从而促使血栓形成和血管堵塞。动物实验也证明,应用TXA2合成酶抑制药可以使缺血/再灌注以后的冠脉血流改善。
二、钙超载
如前所述,钙反常(无钙灌流后用含钙溶液再灌注)时细胞内钙超载,引起严重的功能及结构障碍。钙反常的原因不甚明了,但主要的损伤是在无钙灌流期出现的细胞膜外板(external lamina)与糖被膜(glycocalyx)表面的分离(两者由Ca2+连结在一起)。细胞膜的这种损伤为再灌注时钙的大量内流提供了条件。在长期缺血缺氧后再给氧或再灌注时也可引起细胞内钙超载。
缺血后再灌注时细胞内钙超载的机制尚无定论,但可能与下列因素有关,如图11-1哺乳类钙代谢模式图所示:
图11-1 哺乳类细胞钙代谢模式图
①电压依赖性钙通道;②质膜钙泵ATP酶;③Na+-Ca2+交换;④线粒体;⑤肌浆网;⑥细胞内蛋白或阴离子结合的钙;⑦膜磷脂的极性头部;⑧结合于质膜糖被的钙
(一)钠的平衡障碍 有人证明,再灌注时钙超载是由于钠平衡障碍所致。因为在缺血缺氧时发生了细胞酸中毒,细胞内pH值降低,所以在再灌注时细胞内外形成pH梯度差,由于Na+-H+交换,致细胞内钠增加,然后又依Na+-Ca2+交换机制使细胞外钙大量内流造成细胞钙超载。
(二)细胞膜通透性增高缺血缺氧引起的细胞酸中毒在再灌注时通过细胞内外Na+-H+交换和Na+-Ca2+交换而使细胞内钙增加,而细胞内钙增加可激活磷脂酶,使膜磷脂降解,细胞膜通透性增高,故在灌注时细胞外钙顺着浓度梯度而大量内流,细胞膜通透性增高的更重要的原因可能是再灌注时氧自由基的大量产生。氧自由基可引发细胞膜的脂质过氧化,使膜受损,通透性增高。
(三)线粒体受损 有些学者认为原发性损伤在于线粒体。如所周知,缺血时线粒体结构的功能障碍出现最早,表现为线粒体肿胀、嵴断裂。线粒体膜流动性降低,氧化磷酸化功能受损ATP生成障碍。因此,使上述损伤更为严重。
ATP减少使肌膜及肌浆网膜钙泵功能障碍,由于钙泵功能障碍不能排出和摄取细胞浆中过多的钙,致使细胞浆中游离钙浓度增加而造成钙超载。细胞浆中过多的钙最终形成磷酸盐沉积于线粒体,使线粒体结构及功能更加破坏。
细胞钙超载是再灌注损伤的一个重要特征,但目前,仍未能搞清它究竟是再灌注损伤的原因抑制或结果。
三、白细胞的作用
白细胞(主要是中性粒细胞)出现于梗塞的心肌中已为尸检所证实。1984年Mullane及其同事证明,冠状动脉堵塞60分钟时心肌组织就有白细胞出现,5小时后在缺血区有大量的白细胞聚集。根据Engler及其同事的研究,再灌注时白细胞数非但不减少反而增加。以犬心肌缺血为模型,再灌注仅5分钟,心内膜中性粒细胞就增加25%,缺血轻的组织白细胞聚集也少。
组织缺血和再灌注时白细胞浸润增加的机制还不十分清楚。可能是由于组织受损时,细胞膜磷脂降解,花生四烯酸代谢产物增多,其中有些物质具有很强的趋化作用,因而就能吸引大量白细胞进入组织或粘附于血管内皮,而白细胞本身又能释放很多具有趋化作用的炎性介质,如白三烯之一的LTB4,从而使微循环中白细胞进一步增加。
白细胞积聚对组织的损伤作用在于:
(一)嵌顿、堵塞毛细血管有助于形成无复流现象。微动脉及微静脉亦有大量白细胞粘附于内皮细胞,虽不一定堵塞血流,但粘附的白细胞仍可损伤组织并释放趋化因子从而吸引更多的细胞。
(二)白细胞可以增加血管通透性,水肿组织的含水量与白细胞密度呈正相关,说明白细胞可能引发水肿。白细胞增加血管通透性、引发水肿的机制与白细胞释放的某些炎症介质有关。
(三)激活的中性粒细胞释放溶酶体酶,可使组织发生蛋白水解性破坏和液化。
(四)中性粒细胞可通过产生氧自由基而损伤组织。
白细胞在缺血再灌注损伤的作用,可被以下实验结果证明:
1.用除去白细胞的血液进行再灌注,可以防止水肿产生并减轻再灌性损伤。
2.抗炎药减轻组织白细胞浸泣,可缩小梗塞面积。实验证明布洛芬(ibuprofen)对心肌具有保护作用,主要是由于抑制白细胞浸润的作用所致。
3.用补体抑制药降低补体,从而减少白细胞浸润,可能减轻组织损伤。丝氨酸蛋白酶抑制药,可以缩小心肌梗塞,而这种酶恰是一种在心肌缺血时能激活补体的酶。因此这种酶的抑制药,可通过抑制补体的激活而抑制白细胞浸润,从而减轻心肌损伤。
四、高能磷酸化合物的缺乏
心肌正常情况下以有氧代谢形式生成三磷酸腺苷(ATP)供作功需要。心肌缺血时则转为无氧代谢为主,ATP合成减少,以致心舒缩功能障碍。在犬的实验中证明,心肌严重缺血15分钟(结扎冠状动脉左旋支),心肌发生可逆性损伤。此时如果得到血液再灌注,则细胞并不死亡。但有很多报告指出,短时间缺血后,收缩功能长时间不能恢复。究其原因,多认为与ATP水平的低下有关。研究证明缺血15分钟时不仅ATP减少60%。总腺苷酸池也减少50%。ADP也轻度减少(可能转为ATP或AMP),AMP明显升高,但其升高程度小于ATP减少辐度。再灌注20分钟ATP明显回升,但只接近正常的一半,再灌注24小时仍然维持在低水平上,只有在再灌注4天后ATP及总腺苷池才近于恢复,但仍低于非缺血区(图11-2)。
图11-2 心肌缺血/再灌注时ATP、Ca2+、K+的变化
以大鼠离体作功心脏为模型(Neely氏模型),先在20℃低温下给心脏停跳液,再短时间全心缺血(夹住主动脉)后,再灌注生理溶液。结果是给停跳液30分钟再缺血30分钟后,ATP几乎完全丧失,ADP明显减少,AMP明显增加,总腺苷酸量显著降低。再灌注60分钟可使ATP明显回升,但不及正常对照的一半,而总腺苷酸量则明显低于给停跳液后,比正常对照减少50%。上述研究提示缺血及再灌注损伤的心肌有氧代谢性发生严重损伤,影响能量代谢及心肌功能的恢复。
再灌注时高能磷酸化合物之所以恢复慢且总腺苷酸水平明显下降,可能与下列因素有关。
(一)缺血心肌的代谢障碍主要表现为对氧的利用能力受限,有氧代谢严重受损。在缺血进入不可逆阶段再灌注时,氧的利用并不增加,心肌只能利用运至心肌的氧的17%。氧的利用能力受限与缺血及再灌注所致线粒体受损有关。
(二)ATP合成的前身物质(腺苷、肌苷、次黄嘌呤等)在再灌时被冲洗出去,使心肌失去再合成高能磷酸化合物的物质基础。实验证明在再灌注液中补充肌苷或谷氨酸能促进ATP的合成及心功能的恢复。
3.线粒体膜发生氧自由基诱发的脂质过氧化反应使线体受损。线粒体膜富有磷脂,线粒体在缺氧时又是产生自由基的场所,因此极易引起膜脂过氧化使线粒体功能障碍。
五、自由基的作用
自由基(free radical)是具有一个不配对电子的原子和原子团的总称。由氧诱发的自由基称为氧自由基或活性氧,如超氧阴离子(O2)、羟自由基(OH.)及单线态氧(1O2,激发态放出一个光子)等非脂性自由基。H2O2非自身基,但也是一种氧化作用很强的活性氧。氧自由基与多聚不饱和脂肪酸作用后生成的中间代谢产物烷自由基(L.)、烷氧基(LO.)、烷过氧基(LOO.)等属于脂性自由基。氧自由基和脂性自由基的性质极为活泼,易于失去电子(氧化)或夺取电子(还原),特别是其氧化作用强,故具有强烈的引发脂质过氧化的作用。在生理情况下,氧通常是通过细胞色素氧化酶系统接受4个电子还原成水,同时释放能量,但也有1~2%的氧接受一个电子生成O2,或再接受一个电子生成H2O2。但由于细胞内存有超氧化物歧化酶(superoxide dismutase, SOD)和谷胱甘肽过氧化物酶(glrtathione peroxidase, GSH-PX)等抗氧化酶类可以及时清除它们,所以对机体并无有害影响。在病理条件下,由于活性氧产生的过多或抗氧化酶类活性下降,则可引发链式脂质过氧化反应损伤细胞膜系并进而使细胞死亡。
(一)细胞内氧自由基的生成
分子氧在线粒体细胞色素氧化酶系统中接受一个电子而被还原生成O2。
O2e- → O2-
这是其他活性氧产生的基础。过氧化氢(H2O2)及羟自由基(OH.)续发于此。即氧在获得一个电子时还原生成O2-,获得2个电子生成H2O2,获得3个电子生成OH.,获得四个电子生成H2O。
H2O2既可由O2-自发歧化产生,也可经酶促歧化而生成。
H2O2本身并非自由基而是一种活性氧,它与氧自由基的产生有密切关系(详下文)。
OH.自由基的产生不仅需要O2-或H2O2,而且要有过渡金属,如铁的螯合物的存在。由铁催化的Fenton型Haber-Weiss反应可迅速形成OH.,而单纯的Haber-W-eiss反应速度很慢,很难由此形成OH.。
O2-+H2O2→O2+OH-+OH.
(Haber-Weiss反应)
Fe(Ⅲ)+O2-→fe(Ⅱ)+O2 (2)
Fe(Ⅱ)H2O2 →Fe(Ⅲ)+OH-+OH. (3)
也就是说,O2-使铁还原,还原的铁再使H2O2还原生成OH.,OH.是最活跃最强力的氧自由基。缺血与再灌注时氧自由基生成过多,其机制可能是:
1.黄嘌呤氧化酶的形成增多 黄嘌呤氧化酶(xanthine ocidase, XO)的前身是黄嘌呤脱氢酶(xanthine dehydrogenase, XD)。这两种酶主要存在于毛细血管内皮细胞内。正常时只有10%以XO的形式存在,90%为XD。缺血时由于ATP减少,膜泵功能失灵,Ca2+进入细胞激活Ca2+依赖性蛋白水解酶,使XD大量转变为XO。缺血时ATP不能用来释放能量,而且还依次降解为ADP、AMP和次黄嘌呤,故在缺血组织内次黄嘌呤大量堆积。再灌注时,大量分子氧随血液进入缺血组织,黄嘌呤氧化酶在催化次黄嘌呤转变为黄嘌呤并进而催化黄嘌吟转变为尿酸的两步反应中,都同时以分子氧为电子接受体,从而产生大量的O2-和H2O2,后者再在金属离子参与下形成OH.。因此,再灌注时组织内O2-、OH.等氧自由基大量增加。
2.中性粒细胞 中性粒细胞在吞噬活动时耗氧量显著增加,所摄取的O2绝大部分经细胞内的NADPH氧化酶和NADH氧化酶的作用而形成氧自由基,并用以杀灭病原微生物。如氧自由基产生过多或机体清除氧自由基的酶系统活性不足或抗氧化剂不够时,中性粒细胞形成的氧自由基就可损害组织。
在再灌注时,由黄嘌呤氧化酶的作用所产生的氧自由基起原发的、主要的作用;这些自由基作用于细胞膜后产生的具有趋化活性的物质如LTB4等可吸引大量中性粒细胞到局部释放氧自由基等物质而进一步损害组织。
3.线粒体 可能是由于缺氧使ATP减少,Ca2+进入线粒体增多而使线粒体功能受损,细胞色素氧化酶系统功能失调,以致进入细胞内的氧,经单电子还原而形成的氧自由基增多而经4价还原而形成的水减少。细胞色素氧化酶的功能失调,也可能是缺氧时细胞内氧分压降低的结果。
4.儿茶酚胺的增加 交感-肾上腺髓质系统是机体在应激时的重要调节系统。在各种应激包括缺氧的条件下,此系统分泌大量的儿茶酚胺,儿茶酚胺一方面具有重要的代偿调节作用,但过多的儿茶酚胺特别是它的氧化产物,往往又成为对机体的有害因素。实验证明,大量的异丙肾上腺素、去甲肾上腺素、肾上腺素均能引起细胞损伤。造成心肌损害的是儿茶酚胺的氧化产物,而非儿茶酚胺本身。儿茶酚胺氧化能产生具有细胞毒性的氧自由基。肾上腺素代谢产生紧上腺素红的过程中有O2-产生。
(二)自由基反应与再灌注损伤
机体在生命过程中所能遇到的自由基种类很多。因此很难概括其生物学反应。自由基参与一个反应系统后能形成新的自由基。因此自由基一旦形成,就成为自由基反应扩展程序的一部分,例如:
R.+XH→RH+H.
R.+CCl4→RCl+Cl3C.
另一个自由基反应则是自由基加入到不饱和键中去。如脂肪酸及芳香族环的不饱和键。
自由基反应既可经自由基中间代谢产物不断向前发展,又可由细胞损伤而终止。自由基反应的扩展可以是无限的,但又可为各种自由基清除剂(free radical scavenger)所终止。
由于自由基有极为活泼的反应性,所以它们能和各种细胞成分(膜磷脂、蛋白、核酸)发生反应。
(1)膜脂:是构成膜脂质双层的重要结构及功能成分,富含不饱和脂肪酸,自由基与不饱和脂肪酸作用引发脂质过氧化(lipid peroxidation)反应。脂质过氧化物的形成使膜受体、膜蛋白酶和离子通道的脂质微环境改变,从而改变它们功能,由于脂质过氧化反应的增强,细胞膜内多价不饱和脂肪酸减少,生物膜不饱和脂肪酸/蛋白质比例失常,膜的液态性、流动性改变,通透性增强。含双键脂肪酸过氧化可生成丙二醛,它的产生与脂质过氧化相平行,因而测定丙二醛含量可代表脂质过氧化物的浓度。丙二醛能使膜成分之间形成交联和聚合(polymerization),使膜的基本特性如变构、离子传递、酶活性等发生改变(图11-3)。
图11-3 自由基对膜的损伤
自由基可通过诱致过氧化而影响脂质,从而产生短链脂酰衍化物和副产物
丙二醛。丙二醛反应可介导各种交联反应。自由基也能催化氨基
酸氧化、蛋白质-蛋白质交联和蛋白质链的断裂
(2)蛋白质:在自由基的作用下,胞浆及膜蛋白及某些酶可交联成二聚体或更大的聚合物。这种交联既可借助于蛋白质之间的二硫键形成也可由于自由基损伤的氨基酸残基间的反应形成。蛋白质的交联将使其失去活性,结构改变。
(3)核酸:自由基对细胞的毒性作用主要表现为染色体畸变,核酸碱基改变或DNA断裂。80%是OH.的作用。OH.易与脱氧核糖及硷基反应并使其改变。
(三)细胞对自由基损伤的防护
自由基的产生既然是有机体在正常或病理条件下的常见现象,因此在进化过程中也就形成了一系列对抗自由基,防止其损伤的系统。这种生化学防护系统主要有两大类:低分子自由基清除剂及复合酶系统。
1.低分子清除剂 存在于细胞脂质部分的自由基清除剂有维生素E(α-生育酚)和维生素A(β-胡萝卜素);存在于细胞内外水相中的自由基清除剂有半胱氨酸、抗坏血酸和谷胱甘肽等,它们能提供电子使自由基还原,故有重要的防护作用。例如维生素E(生育酚)能还原O2-、单线态氧(1O2)、过氧化脂质自由基等;抗坏血酸具有相同作用而且能协助生育酚维持其具有活性的还原状态。β-胡萝卜素是单线态氧(1O2)的有效清除剂并能抑制脂质过氧化。
胞浆中的还原型谷胱甘肽(GSH)与还原型辅酶Ⅱ(NADPH)在某些酶如过氧化氢酶、谷胱甘肽过氧化物酶等的协同作用下,能还原H2O2、过氧化脂质、二硫化物及某些自由基。
2.酶性清除剂 细胞内有二种酶可以清除H2O2,即过氧化氢酶和过氧化物酶。如所周知H2O2是OH.自由基的前身,上述两酶可使H2O2浓度降低,从而避免高毒性OH.的产生。这两个酶的细胞内分布,尚不清楚。
细胞内具有清除剂作用的另一个重要酶是超氧化物歧化酶(SOD)。它是一种金属蛋白,可以歧化O2-生成H2O2。哺乳类细胞含有两种SOD。其一是位于胞浆中的CuZn 超氧化物歧化酶,另一是位于线粒体中的Mn超氧化物歧化酶。SOD作用的重要意义,在于清除H2O2及OH.的前身O2-,从而保护细胞不受强毒性氧自由基的损伤。
清除剂的浓度及活性下降必将引起自由基所致之细胞损伤。
(四)自由基在缺血再灌注损伤机制中的地位
缺血/再灌注损伤机制中的各种学说,无一不与自由基的作用有关。
1.缺血/再灌注时脂质过氧化增强(自由基引发),组织及血浆中脂质过氧化物显著增高,超微结构严重受损。给予抗氧化剂如:维生素E、硒(谷胱甘肽过氧化物酶辅基所含金属)及SOD能显著减轻缺血/再灌注损伤。
2.细胞膜脂质过氧化改变膜酶、离子通道的脂质微环境,从而使膜通透性增高,细胞外钙离子内流。膜上Na+-K+-ATP酶失活,可使细胞内Na+升高,Na+-Ca2+交换增强。而使细胞内钙超载。
3.线粒体膜富有磷脂,缺血/再灌注时自由基引发的线粒体膜脂质过氧化或细胞内形成脂质过氧化物作用于线粒体膜,使膜的液态及流动性改变,从而导致线粒体功能障碍,高能磷酸化物产生减少,自由基产生增多。细胞丧失能量贮备。依靠能量的质膜及肌浆网膜钙泵,由于能量不足不能将肌浆中过多的Ca2+泵出或吸收入肌浆网,致使肌细胞内Ca2+浓度增加,加上由细胞外来的Ca2+终于造成细胞内Ca2+超载,成为细胞致死原因。
4.自由基引发的脂质过氧化造成细胞成分间的交联(脂质-脂质交联、蛋白-蛋白交联、脂质-蛋白交联、蛋白-胶原交联),使整个细胞丧失功能。
5.缺血/再灌注时,微粒体及质膜上的脂加氧酶(lipoxygenase)及环加氧酶(cyclooxygenase)激活,催化花生四烯酸代谢,在加强自由基产生及脂质过氧化的同时形成具有高度生物活性的物质,如前列腺素、血栓素等。很多实验证明,缺血特别是再灌注时血栓素形成增加,前列环素形成减少,因而造成微循环障碍,出现无复流现象。
总之,自由基即使不是缺血/再灌注损伤的唯一发病学因素,至少也是甚为重要的环节。
相关热词搜索:
上一篇:缺血/再灌注损伤的治疗
下一篇:心、脑、肠的缺血/再灌注损伤


Syndrome of Insufficient Altitude Adapta")







论坛新帖
频道总排行
医学推广
频道本月排行
热门购物
评论排行
- 2011年临床执业医师考试实践技能真...(13)
- 腋臭手术视频(11)
- 2008年考研英语真题及参考答案(5)
- 节食挑食最伤女人的免疫系统(5)
- 核辐射的定义和单位(5)
- CKD患者Tm与IMT相关(5)
- 齐鲁医院普外科开展“喉返神经监护...(5)
- windows7激活工具WIN7 Activation v1.7(5)
- 正常微循环(5)
- 美大学性教育课来真的 男女上阵亲...(4)