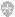膜增生性肾小球肾炎 ——老问题的新面貌
2012-03-25 11:33:51 来源:医学论坛网 作者: 评论:0 点击:
桑吉夫·塞西(Sanjeev Sethi)等 美国明尼苏达州罗彻斯特市梅奥医院病理学与实验室医学科
膜增生性肾小球肾炎(MPGN) 也称为系膜毛细血管性肾小球肾炎,其诊断是依据一组多种多样疾病所共有的一种肾小球损伤模式。MPGN大约占所有活检确诊的肾小球肾炎的 7%~10%1-4,是原发性肾小球肾炎引起的终末期肾病的第三或第四位主要原因2,5。虽然我们对一些与MPGN相关的疾病已经有了很好的了解,但是, 近期的进展还使另外的MPGN相关疾病得以确认。
临床表现
MPGN最常见于儿童,但也可发生在其他任何年龄的人群中。MPGN的临床表现及病程具有高度的变异性,从良性、缓慢进展到迅速进展的范围内表现不 一。因此,患者可以表现为无症状性的血尿与蛋白尿、急性肾炎综合征、肾病综合征、慢性肾脏病,或者甚至是急进性肾小球肾炎。临床表现多种多样是由疾病发病 机制的不同以及与临床病程相关的诊断性活检(进行)的时机不同所致。肾脏损害的程度也呈多样性,可能出现、也可能不出现高血压。在疾病早期就发病、肾脏活 检显示为增生性病变的患者,更可能是肾炎表型,而(活检为)新月体MPGN的患者,可能表现为急进性肾小球肾炎。相反,活检显示同时存在包括修复和硬化的 晚期改变患者,更可能为肾病表型。典型的MPGN患者常常同时具有急性肾炎综合征与肾病综合征的特点,将之称为肾炎-肾病表型 。
分类与病理生理学
光镜下MPGN的典型特点包括系膜细胞过多、毛细血管内增生以及毛细血管壁重塑(形成双轨征)——所有这些均引起肾小球(毛细血管丛)的小叶结构突 出。这些变化是由免疫球蛋白、补体因子或者二者共同在肾小球系膜内或沿肾小球毛细血管壁沉积所致。根据电子显微镜下的所见,在传统上,将MPGN分类为原 发性(特发性)Ⅰ型 MPGN(MPGNⅠ)、Ⅱ型(MPGNⅡ)或Ⅲ型(MPGNⅢ)或继发性MPGN。MPGNⅠ是最常见的类型,以内皮下沉积为特 点,而MPGNⅢ同时出现上皮下以及内皮下沉积6,7。MPGNⅡ以肾小球基底膜内的致密物沉积为特点(“致密物沉积病”)。继发性MPGN(瑞克曾描述 过该病8)常常是由丙型肝炎及其他一些感染所致。
按照目前的分类方法, MPGNⅠ与MPGN Ⅲ可能同时包括免疫复合物介导与补体介导的MPGN病例。鉴于我们最近在认识MPGN中补体替代途径作 用方面所获得的进展,将MPGN视作是免疫复合物介导或补体介导的(疾病)是更为实际的(方法)9。因此,当循环免疫复合物水平升高时,就可能发生免疫复 合物介导的MPGN,而补体介导的MPGN发生的原因是(一些)与补体替代途径失调相关的疾病。
免疫复合物介导的MPGN
抗原血症引起免疫复合物在肾小球的沉积,继而导致免疫复合物介导的MPGN,伴随由慢性感染所致的抗原抗体免疫复合物形成,由自身免疫疾病所致的循环 中免疫复合物水平升高,或者由单克隆γ-球蛋白病所致的病变蛋白血症。免疫复合物触发补体经典途径的激活以及经典途径与终末补体途径的补体因子在系膜内及 沿毛细血管壁的沉积(图1及补充附录中的图1可与本文全文一起在NEJM.org获得)。在免疫荧光显微镜下,肾脏活检标本通常可以见到免疫球蛋白与补 体。

丙型肝炎与其他感染
伴或不伴循环冷球蛋白的慢性病毒感染(如乙型肝炎及丙型肝炎)是MPGN的一个重要原因。20世纪90年代时发现丙型肝炎是免疫复合物介导的MPGN 的一个常见原因,现已将其视为引起MPGN的主要病毒感染8,10-13。除病毒感染外,慢性细菌感染(如心内膜炎、分流性肾炎与脓肿)、真菌感染均与寄 生虫感染与MPGN相关, 在发展中国家尤为如此14-18。与MPGN有关的细菌包括葡萄球菌、结核分枝杆、链球菌、痤疮丙酸杆菌、肺炎支原体、布氏杆 菌、伯纳特立克次体、诺卡菌与脑膜炎双球菌19-30。
自身免疫性疾病
MPGN发生于许多自身免疫性疾病中,主要包括系统性红斑狼疮,偶见于干燥综合征、类风湿关节炎及混合性结缔组织病31-35。
单克隆γ-球蛋白病
近期的研究表明,单克隆γ-球蛋白病 (也称为异常蛋白血症或浆细胞恶性增生)引起的、伴或不伴冷球蛋白的单克隆免疫球蛋白在肾小球的沉积与MPGN 有关36-39。单克隆γ-球蛋白病以合成免疫球蛋白的淋巴细胞或浆细胞单一集落增生为特点,这种增生导致循环中出现单克隆免疫球蛋白。在一项单中心研究 中,研究者采用血清电泳、尿液电泳或二者联用的方法进行评价,发现41%的不伴自身免疫性疾病(过程)或慢性感染的MPGN患者有单克隆γ-球蛋白病证 据 36。这类患者的骨髓活检显示了多种病况:意义不明的单克隆γ-球蛋白病 (MGUS,最常见的情况)、低级别 B细胞淋巴瘤、淋巴浆细胞淋巴瘤、慢 性淋巴细胞白血病及多发性骨髓根据。笔者建议应将患有单克隆γ-球蛋白病 以及MPGN的患者归类为患有“单克隆γ-球蛋白病 相关性MPGN”,而不是 MGUS36。
补体介导的MPGN
补体级联(反应)在天然免疫中具有重要作用。补体因子可诱导强烈炎症反应的发生,引起吞噬细胞的趋化及对细胞(包括微生物)的调理与溶细胞作用。补体 激活通过经典、凝集素或替代途径发生,各途径交汇于C3转化酶,该酶将C3裂解为C3a与C3b。在有B因子与D因子的情况下,C3b与C3 转化酶联 合,进一步生成更多的C3 转化酶,并形成强效的放大环路(补充附录中的图2与图1)。因此,C3 转化酶是补体级联反应中的一个节点。C3b与C3转化 酶的结合,也导致C5转换酶的形成,继而激活终末补体复合物途径以及在细胞表面形成膜攻击复合物(C5b-C9),最终引起细胞的裂解40,41。
替代途径通过C3硫酯键的自发水解在循环中(液相)保持低水平的持续激活(“低速运转”机制 ),生成C3b,随后,C3b与宿主细胞膜、细胞外膜 [如肾小球基底膜(表面相)]以及致病微生物膜结合42,43。为防止自我损害,替代途径的激活受到受精细的调节,并以连续的方式进行。多重补体调节与补 体抑制蛋白作用于级联反应的不同水平,特别是在C3及C5转化酶水平41。这类血浆或液相调节子包括H因子、I因子、H因子相关蛋白1至5以及细胞结合 (cell-bound)和表明调节子[如促衰变因子 (CD55)、补体受体1、CD59、膜辅因子蛋白1(CD46),以及免疫球蛋白超家族补体受体 40-42。H因子加速C3转化酶的衰变,是I因子的辅因子,协助介导的C3b的裂解及失活41,42,从而在液相调控替代途径。终末补体复合物的液相调 节因子包括玻连蛋白与簇集素。某些液相调节因子(包括H因子与H因子相关蛋白1)也附于细胞表面与细胞外膜上,从而增加了一个防止活性补体产物形成的额外 保护机制41。表面调节因子通过使沉积于细胞表面与基底膜的C3b失活来调控C3转化酶44。
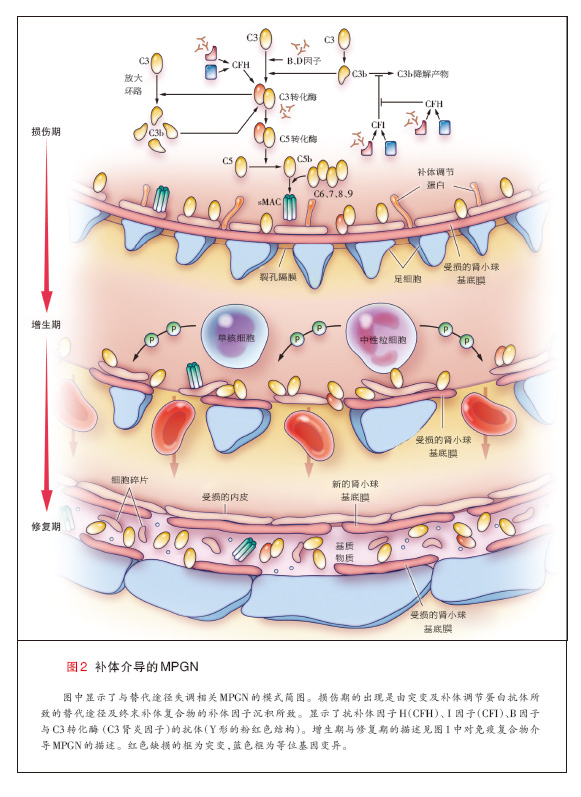
替代途径的失调可以因补体调节蛋白的突变或产生其自身抗体而发生(图3)。例如,如果调节C3转化酶装配与激活以及C3b降解的蛋白(如H、I和B因 子以及H因子相关蛋白5)发生突变,就可导致替代途径的失调45-53。C3本身的杂合子突变导致替代途径的液相失调,这是因为突变的C3可抵抗C3转化 酶的裂解。瘤。 另外,通过低速运转机制所生成的异常的C3转化酶存在C3突变,使其能够抵抗H因子引起的失活。异常的C3转化酶继而裂解正常C3等位基 因所产生的C3,导致C3降解产物水平增加54。同样,抗补体调节蛋白(如H、B因子)抗体及抗C3转化酶本身的抗体,可导致替代途径的过度激活 49,55。抗C3转化酶抗体(称为C3肾炎因子)使转化酶稳定,并通过预防转化酶的失活和降解来延长其半衰期,从而激活替代途径40,45。
H与B因子、膜辅因子蛋白及C3的某些基因多态性也与MPGN相关56,57。编码H因子基因的多态性(特别是Tyr402His等位基因变异)是研 究最多的基因多态性。与Tyr402相比,His402在MPGN及替代途径异常患者中出现过多,功能研究显示,His402损害H因子介导的细胞表面 C3转化酶的调节49,58,59。
无论是何种机制,替代途径的失调导致活性补体产物的生成,包括C3b与终末补体因子,它们被任意转运至内皮表面(包括肾小球)41,60。这些补体产 物与碎片在系膜与内皮下区域的沉积引发肾小球炎症,并导致MPGN。免疫球蛋白不直接参与MPGN的致病,因此,在荧光显微镜下,补体介导的MPGN通常 为免疫球蛋白阴性而补体阳性。
尽管存在多种遗传危险因素,补体异常引起的MPGN常常在一生中相对较晚的时候发病,提示其发病要求有另外的损伤或环境因素存在。此外,在高危家系 中,并非所有基因相似的成员都发生MPGN,这再次表明其发病需要其他疾病诱导因素的存在。我们推测,当存在导致对疾病易感的突变或者等位基因变异而不发 生MPGN时,可能(是因为)存在充分(足够)的调节机制43。但是,当有另外的损伤(如补体激活的感染)发生时,就可能会击溃这种代偿性的调节机制,触 发补体因子在肾小球的沉积41。这种情况可能可以解释在许多MPGN患者中所发现的与感染有关的反复发作的肉眼血尿(咽炎同步血尿)。同样,另外的损伤 (例如,MGUS患者中作为抗补体调节蛋白自身抗体的单克隆蛋白的产生)可导致替代途径的失调以及MPGN的发病61,62。
病理特点
免疫球蛋白、补体或二者同时在毛细血管壁的系膜及内皮下区域的沉积引发急性损伤,随后常常是炎性(细胞性的或增生性的)期,伴有炎性细胞内流。接下来 是修复期,在该期内,新出现的系膜基质导致系膜扩张,同时新的小球基底膜生成,这些膜看上去像是复制的基底膜[称为双轨征(tram tracks或 double contours)](图1与图2)。
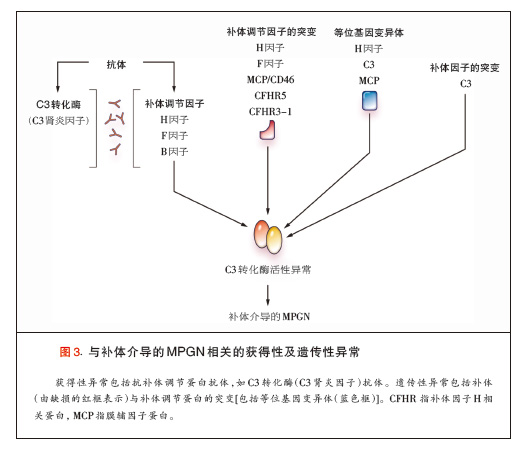
将免疫荧光结果用于区分免疫复合物介导的MPGN与补体介导的MPGN,并常常指向某一个特定的病因。例如,与单克隆γ-球蛋白病相关的MPGN显示 κ或λ轻链限制性的单型免疫球蛋白(图4)。与丙型肝炎病毒感染相关的MPGN常常显示IgM、IgG、C3与κ及λ轻链。伴自身免疫性疾病的 MPGN 模式常常包括多种免疫球蛋白与补体蛋白——IgG、IgM、IgA、C1q、C3及κ与λ轻链。与替代途径功能障碍相关的MPGN以系膜内及沿 毛细血管壁的明亮的C3免疫染色为特点(图4)。免疫荧光显微镜下没有显著的免疫球蛋白染色,(有助于)从免疫复合物介导的MPGN中区分出替代途径功能 障碍引起的MPGN。
电子显微镜常常显示系膜与内皮下的沉积物,在一些病例中,还可显示膜内与上皮下的沉积物。修复期期间,在新基底膜物质内,新基底膜形成,毛细血管壁内 沉积,伴有来源于炎性、系膜和内皮细胞的细胞成份,其结果是毛细血管壁增厚及出现沿毛细血管壁的双轨征。除致密物沉积病外,电子显微镜无法区分免疫复合物 介导的MPGN与补体介导的MPGN。电子显微镜下的表现,可将替代途径失调导致的MPGN进一步分为致密物沉积病与C3肾小球肾炎(C3GN)。致密物 沉积病的特点是嗜锇性、腊肠样、波浪状的致密沉积物替代肾小球基底膜,也发生于系膜内,而C3GN是有系膜、内皮下(有时为上皮下及膜内)的沉积物(图 4)。根据电子显微镜下的C3GN形态学特征,按照旧的分类,C3GN最可能被称为MPGNⅠ或MPGNⅢ。从患C3GN的患者中获得的肾小球激光显微切 割及质谱分析数据与替代途径的非限制性激活一致,而这类患者中的蛋白质组学谱与致密物沉积病患者相似49,60。有研究者假设,致密物沉积病与C3GN是 一个连续疾病的一部分,该假说得到某些病例的进一步支持,这些病例表现出的特征是介于致密物沉积病与C3GN之间的状态,在电子显微镜下,某些毛细血管袢 显示的是致密物沉积病的腊肠样膜内沉积,而其他毛细血管袢显示的是C3GN的内皮下与上皮下沉积。
在补充附录中,讨论了5例具有明确的可识别病因的免疫复合物介导或补体介导的MPGN(患者)。
替代途径失调与疾病亚型
替代途径失调在某些患者中引起致密物沉积病,而在其他患者中引起C3GN,最可能的原因是功能失调的程度及位点(或二者均有)存在差异。另外,补体调节蛋白的某些等位基因变异可能与致密物沉积病有关,而其他变异可能与C3GN有关57。
免疫复合物介导与补体介导的肾小球损伤的其他模式
除MPGN外的其他肾小球损伤模式可能是由免疫球蛋白、补体(或二者同时)沉积所引起的。例如,系膜增生性肾小球肾炎、弥漫增生性肾小球肾炎、新月体 肾小球肾炎及硬化性肾小球病均可能同时出现在C3GN与致密物沉积病中63,64。“C3肾小球病”是一种泛称,它描述了损伤的不同模式65,这一病变可 能取决于多种因素,包括损伤的严重程度以及实施活检时的疾病(过程)时相(急性或者慢性),以前的治疗也可能影响活检的结果。
无免疫复合物或补体的MPGN
在内皮细胞损伤导致的血栓形成性微血管病中也发现了与MPGN一致的损伤模式。急性期时,肾小球毛细血管内出现肾小球膜溶解、内皮肿胀及纤维栓塞。病 变(过程)进展为修复期及慢性期时,发生系膜扩张与肾小球毛细血管壁重塑(包括双轨征形成)。因此,血栓性血小板减少性紫癜或溶血尿毒症综合征的愈合期、 与补体异常有关的不典型的溶血性尿毒性综合征、抗磷脂抗体综合征、药物诱导的血栓形成性微血管病、骨髓移植相关性肾病、放射性肾炎、恶性高血压及结缔组织 病均可在活检下表现为MPGN的损伤模式66,67。血栓形成性微血管病中,免疫荧光下常常见不到免疫球蛋白与补体,而电子显微镜下系膜内或沿毛细血管壁 也无电子致密沉积物。
评估
血清补体C3、C4水平持续降低(或二者同时降低)常见于MPGN患者中,C3与C4补体水平均降低在免疫复合物介导的MPGN中更为常见,而C3水平降低、C4水平正常在替代途径功能障碍时更为常见(尤其是急性相时),C3水平正常无法除外替代途径功能障碍。
上一篇:单孔后腹腔镜肾根治切除安全可行
下一篇:双膦酸盐或改善肾细胞癌骨转移预后










论坛新帖
频道总排行
医学推广
频道本月排行
热门购物
评论排行
- 2011年临床执业医师考试实践技能真...(13)
- 腋臭手术视频(11)
- 2008年考研英语真题及参考答案(5)
- 节食挑食最伤女人的免疫系统(5)
- 核辐射的定义和单位(5)
- CKD患者Tm与IMT相关(5)
- 齐鲁医院普外科开展“喉返神经监护...(5)
- windows7激活工具WIN7 Activation v1.7(5)
- 正常微循环(5)
- 美大学性教育课来真的 男女上阵亲...(4)